STAT433 Nonparametric Statistics
SZ
3/17/2022
- Chapter 1. Introduction
- Chapter 2. Bootstrap Confidence Intervals and Randomization Methods for Testing Hypotheses
- The Big Idea of Bootstrap
- Bootstrapping Confidence Interval for One Population Mean
- Bootstrapping CI for the Difference between Two Population Means Based on Independent Samples
- Bootstrap Method for Multiple Regression
- A Look at Hypothesis Tests Based on Parametric Methods
- Obtaining the P-value for the Test about One Population Mean with Resampling
- Obtaining the P-value for the Test about Two Population Means Based on Independent Samples with Resampling
- Bootstrapping Confidence Interval for Correlation
- Obtaining P-value for the Test about Correlation with Resampling
- Chapter 3. Sign Test
- Chapter 4. Wilcoxon Signed-Rank Test
- Chapter 5. Wilcoxon Rank-Sum Test
- Chapter 6. Kruskal-Wallis Test (or H Test)
- Chapter 7. Friedman Test
- Chapter 8. Rank Correlation Tests
- Chapter 9. K-S & A-D Tests for Goodness of Fit
- Chapter 10. Survival Analysis
- References
- Mathematical Foundation to Non-Parametric Statistics
- Readings
- Appendix
Chapter 1. Introduction
Nonparametric statistics is a pretty old (> 120 years) branch of statistics. Its basic theory is very mature. Nonparametric methods are mainly developed for the use with small samples. For large samples, the methods are equally good as parametric methods based on normality.
Compared to parametric tests, nonparametric tests have several advantages, including
Higher power (when assumptions for the parametric methods are violated)
Fewer assumptions
Wide range of use (for all data types, including nominal, interval, or data that have outliers or that have been measured imprecisely)
The disadvantages are:
- Less powerful than parametric tests if assumptions for parametric methods are not violated.
We do need some basic probability theory before starting the introduction to nonparametric methods.
Some classic nonparametric methods are:
Review of Basic Statistics
Experiment, Sample Space, Events, and Probability
An experiment refers to any process of observation or measurement. The collection/set of all possible outcomes of an experiment is called the sample space.
An event is the collection/set of some of the possible outcomes. In other words, an event is a subset of the sample space.
For an experiment with n possible outcomes, if each possible outcome is equally likely, then each outcome occurs with probability of \(\frac{1}{n}\). An event occurs when any of the outcomes in it occurs. The probability that an event occurs is given by \(\frac{k}{n}\), where \(k\) is the number of outcomes in the event.
Examples:
Throw a six-sided fair die only once and record the outcome. The sample space, denoted by \(S\), is \(S=\{1,2,3,4,5,6\}\). The set \(S\) has 64 possible subsets and they are \(\{~\}, \{1\}, \{2\}, \cdots, \{1,2,3,4,5,6\}\). They all correspond to different events. The probability of the event \(\{~\}\) is 0. The probability of the event \(\{1,2,3,4,5,6\}\) is 1. The probability of the event \(\{1, 3, 5\}\) is \(\frac{3}{6}\) or 0.5. What is the probability of getting a prime number? A prime number is a positive integer that has exactly two divisors.
Throw a six-sided fair die twice and record the sum of the two numbers observed. What is the probability of getting the sum of 9? The possible outcomes of the sum are \(2, 3, \cdots, 12\), so the sample space is \(\{2, 3, \cdots, 12\}\). Is the desired probability \(\frac{1}{11}\)? No! What makes our method fail is that the sample space contains outcomes that are not equally likely.
Let’s use a new sample space for (b): \[S=\{(1, 1), (1, 2), (1, 3), (1, 4), (1, 5), (1, 6), \cdots, (6, 1), (6, 2), (6, 3), (6, 4), (6, 5), (6, 6) \}\].
If we collect the outcomes \((3,6), (4,5), (5, 4), (6,3)\) and form a set, called \(A\), then the set is an event, which is equivalent to the event that the sum of the two observed numbers is 9 under the previous sample space. The new sample space has 36 equally likely possible outcomes, so the probability of \(A\) is \(\frac{4}{36}\) or \(\frac{1}{9}\), which is the desired probability.
Random Variables and Their Distributions
A random variable is a variable that takes values depending on the outcomes of a random experiment.
Examples of random variables:
Toss a six-sided fair die only once and record the number of points showing up by \(X\). \(X\) is a random variable.
Toss a six-sided fair die twice and record the total number of points by \(Y\). \(Y\) is a random variable, and it takes values \(2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12\).
Suppose that a population has 3 numbers: 3, 4, and 3. Select 3 numbers from this population with replacement. Denote the mean of this sample by \(\bar{x}\), which is a random variable.
If we list the possible values of a random variable along with the corresponding probability, we have the distribution of the random variable.
The distributions of the above random variables are given below.
D=data.frame(X = c("$x$", 1, 2, 3, 4, 5, 6)
)
D$p = c("probability", rep("$\\frac{1}{6}$", 6))
names(D) = NULL
kable(t(D), align='ccccccc',
caption = "<center><strong>Probability Distribution \nWhen Tossing a Fair Die Only Once</strong></center>",
table.attr = "style='width:60%; '") %>%
footnote("")%>%
kable_classic(full_width = TRUE, position = "center") %>%
column_spec (1:7,border_left = TRUE, border_right = TRUE) %>%
kable_styling("bordered") # Options: basic, striped, bordered, hover, condensed, responsive and none.| \(x\) | 1 | 2 | 3 | 4 | 5 | 6 |
| probability | \(\frac{1}{6}\) | \(\frac{1}{6}\) | \(\frac{1}{6}\) | \(\frac{1}{6}\) | \(\frac{1}{6}\) | \(\frac{1}{6}\) |
| Note: | ||||||
| \(x\) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| \(probability\) | \(\frac{1}{36}\) | \(\frac{2}{36}\) | \(\frac{3}{36}\) | \(\frac{4}{36}\) | \(\frac{5}{36}\) | \(\frac{6}{36}\) | \(\frac{5}{36}\) | \(\frac{4}{36}\) | \(\frac{3}{36}\) | \(\frac{2}{36}\) | \(\frac{1}{36}\) |
| Note: | |||||||||||
The distribution of the random variable in part (c) is found as follows:
- First, label the 3 numbers by \(x_1, x_2\), and \(x_3\). With replacement, the 27 possible samples using the labels are listed in the following table.
| Sample | sample | Sample Mean | Probability |
| \(x_1, x_1, x_1\) | \(3, 3, 3\) | 3 | \(\frac{1}{27}\) |
| \(x_1, x_1, x_2\) | \(3, 3, 4\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_1, x_1, x_3\) | \(3, 3, 3\) | 3 | \(\frac{1}{27}\) |
| \(x_1, x_2, x_1\) | \(3, 4, 3\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_1, x_2, x_2\) | \(3, 4, 4\) | \(\frac{11}{3}\) | \(\frac{1}{27}\) |
| \(x_1, x_2, x_3\) | \(3, 4, 3\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_1, x_3, x_1\) | \(3, 3, 3\) | 3 | \(\frac{1}{27}\) |
| \(x_1, x_3, x_2\) | \(3, 3, 4\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_1, x_3, x_3\) | \(3, 3, 3\) | 3 | \(\frac{1}{27}\) |
| \(x_2, x_1, x_1\) | \(4, 3, 3\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_2, x_1, x_2\) | \(4, 3, 4\) | \(\frac{11}{3}\) | \(\frac{1}{27}\) |
| \(x_2, x_1, x_3\) | \(4, 3, 3\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_2, x_2, x_1\) | \(4, 4, 3\) | \(\frac{11}{3}\) | \(\frac{1}{27}\) |
| \(x_2, x_2, x_2\) | \(4, 4, 4\) | 4 | \(\frac{1}{27}\) |
| \(x_2, x_2, x_3\) | \(4, 4, 3\) | \(\frac{11}{3}\) | \(\frac{1}{27}\) |
| \(x_2, x_3, x_1\) | \(4, 3, 3\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_2, x_3, x_2\) | \(4, 3, 4\) | \(\frac{11}{3}\) | \(\frac{1}{27}\) |
| \(x_2, x_3, x_3\) | \(4, 3, 3\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_3, x_1, x_1\) | \(3, 3, 3\) | 3 | \(\frac{1}{27}\) |
| \(x_3, x_1, x_2\) | \(3, 3, 4\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_3, x_1, x_3\) | \(3, 3, 3\) | 3 | \(\frac{1}{27}\) |
| \(x_3, x_2, x_1\) | \(3, 4, 3\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_3, x_2, x_2\) | \(3, 4, 4\) | \(\frac{11}{3}\) | \(\frac{1}{27}\) |
| \(x_3, x_2, x_3\) | \(3, 4, 3\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_3, x_3, x_1\) | \(3, 3, 3\) | 3 | \(\frac{1}{27}\) |
| \(x_3, x_3, x_2\) | \(3, 3, 4\) | \(\frac{10}{3}\) | \(\frac{1}{27}\) |
| \(x_3, x_3, x_3\) | \(3, 3, 3\) | 3 | \(\frac{1}{27}\) |
| Note: | |||
When focusing on sample means, we can reduce the table to get
| Sample Mean | Probability |
| 3 | \(\frac{8}{27}\) or 0.296 |
| \(\frac{10}{3}\) | \(\frac{12}{27}\) or 0.444 |
| \(\frac{11}{3}\) | \(\frac{6}{27}\) or 0.222 |
| 4 | \(\frac{1}{27}\) or 0.037 |
| Note: | |
Chapter 2. Bootstrap Confidence Intervals and Randomization Methods for Testing Hypotheses
The sampling distribution of a sample statistic (such as sample mean or sample proportion) can be hard to find. In practice, we can use resampling to get an approximation to the sampling distribution.
The Big Idea of Bootstrap
The idea of bootstrap is this:
 The above is from: https://online.stat.psu.edu/stat555/node/119/
The above is from: https://online.stat.psu.edu/stat555/node/119/
Bootstrapping Confidence Interval for One Population Mean
The idea is to get many bootstrap samples from the original sample. For each of such bootstrap samples, calculate the sample mean (called the bootstrap statistic in general). The distribution of these bootstrap sample means, called the bootstrap distribution, can be used as an approximation to the true sampling distribution of the original sample mean.
Example 1.
Suppose a sample containing 3, 3, and 4 is obtained from a population. Use the bootstrap method to approximate the sampling distribution of the sample mean.
If there are too many possible bootstrap samples, we can use R to approximate the bootstrap distribution.
# Check the 3rd distribution
N = 5000
M=c() # A NULL vector to be populated with values
for (i in 1:N){
x = c(3, 3, 4)
M[i] = mean(sample(x, 3, replace = TRUE))
}
T = table(M)/N
# Create pretty table for T
D = cbind(as.numeric(names(T)), as.numeric(T))
D = rbind(c("$x$", "$frequency$"), D)
names(D) = NULL
kable(D, align='cc',
caption = "<center><strong>Approximated Bootstrap Distribution <br> of the <span style = 'color:red'>Sample Mean</span></strong></center>",
table.attr = "style='width:60%; '") %>%
footnote("")%>%
kable_classic(full_width = TRUE, position = "center") %>%
column_spec (1:2,border_left = TRUE, border_right = TRUE) %>%
kable_styling("bordered") # Options: basic, striped, bordered, hover, condensed, responsive and none.| \(x\) | \(frequency\) |
| 3 | 0.2904 |
| 3.33333333333333 | 0.4562 |
| 3.66666666666667 | 0.2154 |
| 4 | 0.038 |
| Note: | |
Instead of above, we can use the package “boot” to do bootstrapping.
myData = c(3, 3, 4)
mydata <- data.frame(x = myData) # Create a DATA FRAME with the sample as the only column
# Define the function which calculates the bootstrap statistic for each bootstrap sample
meanfun <- function(data, i){ # i is a vector of n random indices between 1 and n, inclusive
# where n equals the original sample size, which is 3 here
d <- data[i, ]
return(mean(d))
}
library(boot)
bo <- boot(mydata, statistic=meanfun, R=5000) # R bootstrap samples to be generated, each of size n = 3
#bo$t[1:50] # The first 50 of All the R bootstrap statistics
T = table(bo$t)/5000
D = T%>% data.frame()
names(D)[1] = c("$x$")
kable(D, align='cc',
caption = "<center><strong>Approximated Bootstrap Distribution <br> of the <span style = 'color:red'>Sample Mean</span></strong></center>",
table.attr = "style='width:60%; '") %>%
kable_classic(full_width = TRUE, position = "center") %>%
column_spec (1:2,border_left = TRUE, border_right = TRUE) %>%
kable_styling("bordered") # Options: basic, striped, bordered, hover, condensed, responsive and none.| \(x\) | Freq |
|---|---|
| 3 | 0.2988 |
| 3.33333333333333 | 0.4456 |
| 3.66666666666667 | 0.2166 |
| 4 | 0.0390 |
If we want a bootstrap confidence interval for the population mean, we can do
bc = boot.ci(bo, conf=0.95, type="bca") # A confidence interval for the population mean is
# calculated using the R bootstrap statistics with
# the bias-corrected and accelerated (BCa) method
# Other types such as "perc" means the percentile CI.
bc## BOOTSTRAP CONFIDENCE INTERVAL CALCULATIONS
## Based on 5000 bootstrap replicates
##
## CALL :
## boot.ci(boot.out = bo, conf = 0.95, type = "bca")
##
## Intervals :
## Level BCa
## 95% ( 3.000, 3.667 )
## Calculations and Intervals on Original Scalepar(margin = c(4, 4, 7, 4))## Warning in par(margin = c(4, 4, 7, 4)): "margin" is not a graphical parameterhist(bo$t, probability = TRUE,
main = "Probability Histogram of Bootstrap Statistics",
xlab = "Bootstrap Statistics")
abline(v=bc$t0, col = "red", lty = "dashed")
abline(v=bc$bca[4:5], col = "blue", lty = "dashed")
legend(3.5, 4, legend=c('Bias-corrected and accelerated CI', 'Sample mean'), lty=c(2, 2), col=c('blue', 'red'), text.col = c('blue', 'red'), bty = "n")
The above graph only has a few bars, which is due to the fact that we have a very small sample. Try the following data instead:
- myData = c(106, 82, 94, 111, 84, 79, 113, 103, 91, 95, 105, 71, 125, 104, 116, 100, 107, 101, 82, 85)
Bootstrapping CI for the Difference between Two Population Means Based on Independent Samples
When there are two independent samples, one from each quantitative population, we can use the bootstrap to construct a confidence interval for the difference between the means of the two populations.
Since there are two separate samples, we need to do bootstrapping separately for each sample. Here is how we do bootstrapping:
 Modified from this source: https://towardsdatascience.com/introduction-to-bootstrapping-in-data-science-part-2-ef7236e464a
Modified from this source: https://towardsdatascience.com/introduction-to-bootstrapping-in-data-science-part-2-ef7236e464a
Let’s assume that our two samples are:
Sample 1: 34, 45, 28, 40, 32
Sample 2: 8, 16, 9, 10
Here is the R code:
x = c(34, 45, 28, 40, 32)
y = c(8, 16, 9, 10)
R = 5000
bs.diff = c()
for (i in 1:R){
bs.x = sample(x, replace = TRUE) # Generate a bootstrap sample from the first sample
bs.y = sample(y, replace = TRUE) # Generate a bootstrap sample from the second sample
bs.diff[i] = mean(bs.x) - mean(bs.y)
}
hist(bs.diff)
quantile(bs.diff, prob = c(0.025, 0.975)) # Get the percentile confidence interval## 2.5% 97.5%
## 19.20 31.15# Let's use the traditional 2-sample t confidence interval method
t.test(x, y, conf.level = 0.95)##
## Welch Two Sample t-test
##
## data: x and y
## t = 7.1516, df = 6.2968, p-value = 0.0003029
## alternative hypothesis: true difference in means is not equal to 0
## 95 percent confidence interval:
## 16.57609 33.52391
## sample estimates:
## mean of x mean of y
## 35.80 10.75The above two methods give similar confidencee intervals.
We can also use the “boot” package to bootstrapping. We need to create a data frame with one column being the values and the other being groups.
D = data.frame(x=c(34, 45, 28, 40, 32, 8, 16, 9, 10), g = c(1, 1, 1, 1, 1, 2, 2, 2, 2))Find the mean of each sample:
# Two ways to find group means
aggregate(x~g, D,mean) # way 1## g x
## 1 1 35.80
## 2 2 10.75with(D, tapply(x, g, mean)) # way 2## 1 2
## 35.80 10.75We now define the function for calculating the difference between bootstrap means:
# Define function that calculates the statistic
# The first argument is the data and the second is (stratified) resampling of indices.
# Why stratified? This is because two separated resampling is done within each sample.
# The first sample corresponds to indices 1, 2, 3, 4, and 5.
# The second sample corresponds to indices 6, 7, 8, and 9.
# A resampling of the first set of indices might be 2, 5, 2, 1, 1 and
# the second set of indices might be 8, 8, 6, 9. In this case, i is the vector 2, 5, 2, 1, 1, 8, 8, 6, 9.
stats = function(d, i) {
resampledData = d[i, ] # Treat i as a vector representing a resampling of all row indices of data
resampledData1 = subset(resampledData, g == 1)
resampledData2 = subset(resampledData, g == 2)
m1 = mean(resampledData1$x)
m2 = mean(resampledData2$x)
m = m1 - m2
m
}
# Or more concisely
stats2 =function(d, i){
resampledData = d[i, ]
m <- tapply(resampledData$x, resampledData$g, mean)
m[1]-m[2]
}
b = boot(D, stats, R = 500, strata = D$g) # Stratified re-sampling
boot.ci(b, type=c("perc", "bca"))## BOOTSTRAP CONFIDENCE INTERVAL CALCULATIONS
## Based on 500 bootstrap replicates
##
## CALL :
## boot.ci(boot.out = b, type = c("perc", "bca"))
##
## Intervals :
## Level Percentile BCa
## 95% (19.33, 31.30 ) (19.03, 31.18 )
## Calculations and Intervals on Original ScaleBootstrap Method for Multiple Regression
Bootstrap confidence intervals can be made for all coefficients plus any other parameters. Read the reference: https://www.statmethods.net/advstats/bootstrapping.html and https://artowen.su.domains/courses/305a/FoxOnBootingRegInR.pdf
library(boot)
# Step 1: Create a function that obtains bootstrap statistics (regression coefficients
# plus the r-square.)
bs <- function(formula, data, i) {
d <- data[i,] # allows boot to select sample
# Fit a multiple regression model using the lm() function in base R
fit <- lm(formula, data=d)
# Generate a summary of the fit
s <- summary(fit)
# Only need the coefficients and the R-square
return(c(coef(fit), s$r.squared))
}
# Step 2: bootstrapping with 5000 replications and
# calculating the 5000 bootstrap statistics
results <- boot(data=mtcars,
statistic=bs,
R=5000,
formula=mpg~wt+disp # This is needed since the function created
# before needs a formula to fit a specific model
# If you check the detail of the boot() function,
# you will see "..." in the list of its parameters.
# "..." indicates other named arguments for statistic # function created before.
)
# Step 3: view results. Are the bootstrap distributions for each statistic symmetric?
# If yes, a percentile CI is good enough; if not, a bca CI might be needed.
plot(results, index=1) # intercept
plot(results, index=2) # wt
plot(results, index=3) # disp
plot(results, index=4) # R-square
# Step 4: get 95% confidence intervals, one for each parameter
boot.ci(results, type="bca", index=1) # for intercept## BOOTSTRAP CONFIDENCE INTERVAL CALCULATIONS
## Based on 5000 bootstrap replicates
##
## CALL :
## boot.ci(boot.out = results, type = "bca", index = 1)
##
## Intervals :
## Level BCa
## 95% (29.67, 39.73 )
## Calculations and Intervals on Original Scaleboot.ci(results, type="bca", index=2) # for coefficint of wt## BOOTSTRAP CONFIDENCE INTERVAL CALCULATIONS
## Based on 5000 bootstrap replicates
##
## CALL :
## boot.ci(boot.out = results, type = "bca", index = 2)
##
## Intervals :
## Level BCa
## 95% (-5.604, -0.965 )
## Calculations and Intervals on Original Scaleboot.ci(results, type="bca", index=3) # for coefficint of disp## BOOTSTRAP CONFIDENCE INTERVAL CALCULATIONS
## Based on 5000 bootstrap replicates
##
## CALL :
## boot.ci(boot.out = results, type = "bca", index = 3)
##
## Intervals :
## Level BCa
## 95% (-0.0339, -0.0003 )
## Calculations and Intervals on Original Scaleboot.ci(results, type="bca", index=4) # for R-square## BOOTSTRAP CONFIDENCE INTERVAL CALCULATIONS
## Based on 5000 bootstrap replicates
##
## CALL :
## boot.ci(boot.out = results, type = "bca", index = 4)
##
## Intervals :
## Level BCa
## 95% ( 0.6379, 0.8551 )
## Calculations and Intervals on Original Scale# To get CI for all parameters
boot.ci(results, type=c("perc", "bca"))## BOOTSTRAP CONFIDENCE INTERVAL CALCULATIONS
## Based on 5000 bootstrap replicates
##
## CALL :
## boot.ci(boot.out = results, type = c("perc", "bca"))
##
## Intervals :
## Level Percentile BCa
## 95% (30.18, 40.12 ) (29.67, 39.73 )
## Calculations and Intervals on Original ScaleA Look at Hypothesis Tests Based on Parametric Methods
Statistical inference methods include tests of hypotheses and confidence intervals. We review the test of hypotheses.
| Parametric Method | Sample | Assumption | Reference | R code |
| 1-sample t-test for mean | One sample | normality | t.test(x, mu=mu0, alternative=c(‘two.sided’, ‘less’, ‘greater’)) | |
| 2-sample t-test for mean difference | Two independent samples | normality | t.test(x,y, mu=0, alternative=c(‘two.sided’, ‘less’, ‘greater’)) | |
| 1-way ANOVA | The response variable is quantitative and the explanatory variable is categorical | normality | http://www.sthda.com/english/wiki/one-way-anova-test-in-r | aov(y ~ x, data = my_data); pairwise.t.test(my_data\(y, my_data\)x, p.adjust.method = ‘bonferroni’) |
| t-test for correlation | Two quantitative variables | normality | cor.test(x, y, method=c(‘pearson’, ‘kendall’, ‘spearman’)) | |
| Note: | ||||
Obtaining the P-value for the Test about One Population Mean with Resampling
Suppose we have a sample \(x_1, x_2, \cdots, x_n\) from a quantitative population. Consider the problem of testing \(H_0: \mu=\mu_0\) vs. \(H_a: \mu\ne\mu_0\) using \(\bar{x}\) as the test statistic.
When calculating the \(p\)-value, we need to assume the null hypothesis to be true. When using a resampling method, resamples are generated from the modified sample: \(x_1-\bar{x}+\mu_0, x_2-\bar{x}+\mu_0, \cdots, x_n-\bar{x}+\mu_0\). That is, the new sample is obtained by centering the original sample and adding the null value to the centered values. So, this resampling method should be called randomization rather than bootstrap.
Let’s say our data are 10, 14, 21, 25, 30, and 44. The sample mean is 24. We want to test \(H_0: \mu=20\) vs. \(H_a: \mu\ne 20\).
x = c(10, 14, 21, 25, 30, 44)
mu0 = 20
new.x = x - mean(x) + mu0
R = 5000
M = c()
for (i in 1:R){
s = sample(new.x, replace = TRUE) # Generate a bootstrap sample
M[i] = mean(s) # Calculate the mean of the bootstrap sample
}
hist(M, main = "The Histogram of The Means of Randomization Samples", xlab = "Randomization Mean") The randomization distribution should be centered around \(\mu_0\).
The randomization distribution should be centered around \(\mu_0\).
To calculate the \(p\)-value, we count how many resample means are farther away from \(\mu_0\) than the sample mean is, and then divide the result by the number of resamples. So, we do
(sum(abs(M-mu0)>abs(mean(x)-mu0)))/R## [1] 0.3738as shown by the graph below:
hist(M, main = "The Distribuion of Resample Means")
abline(v=c(mean(x), 2*mu0-mean(x)), lty = "dashed", col = c("red", "blue"))
legend("topright", legend = c("mean(x)", "2mu0 - mean(x)"), lty = 2, col = c("red", "blue"), text.col = c("red", "blue"), bty = "n")
The \(p\)-value based on the traditional one-sample t test is
t.test(x, mu = mu0, alternative = "two.sided")##
## One Sample t-test
##
## data: x
## t = 0.8043, df = 5, p-value = 0.4577
## alternative hypothesis: true mean is not equal to 20
## 95 percent confidence interval:
## 11.21582 36.78418
## sample estimates:
## mean of x
## 24If we want to test \(H_0: \mu=20\) vs. \(H_a: \mu > 20\), the \(p\)-value would be equal to the proportion of resample means greater than the original sample mean:
(sum(M > mean(x)))/R## [1] 0.1824as shown by the graph below:
hist(M, main = "The Distribuion of Resample Means")
abline(v=mean(x), lty = "dashed", col = "red")
legend("topright", legend = c("Sample Mean"), lty = 2, col = c("red"), text.col = c("red"), bty = "n")
The \(p\)-value based on the traditional one-sample t test is
t.test(x, mu = mu0, alternative = "greater")##
## One Sample t-test
##
## data: x
## t = 0.8043, df = 5, p-value = 0.2289
## alternative hypothesis: true mean is greater than 20
## 95 percent confidence interval:
## 13.97864 Inf
## sample estimates:
## mean of x
## 24Obtaining the P-value for the Test about Two Population Means Based on Independent Samples with Resampling
When testing hypotheses about the mean difference of two population, traditionally use the 2-sample t test.
We can also use a resampling method to find the p-value based on the difference between sample means.
The method should be rather called randomization than bootstrap. There are three methods available, as explained by https://online.stat.psu.edu/stat200/lesson/5/5.3/5.3.1. We will use the reallocate method (or called permutation method), which usually is the default method. Assume the sample sizes are \(n_1\) and \(n_2\). The steps of the method is:
Step 1. Mix the two samples.
Step 2. Randomly choose \(n_1\) values from the mix and calculate the sample mean. Calculate the mean for the remaining values. Subtract the two means.
Repeat the two steps many times.
The distribution of these differences gives an approximation to the sampling distribution of the original mean difference, under the assumption that the two population means are equal.
The \(p\)-value depends on the alternative hypothesis.
If the alternative hypothesis is left-tailed, the \(p\)-value equals the proportion of the differences that are less than the difference in the original sample means.
If the alternative hypothesis is right-tailed, the \(p\)-value equals the proportion of the differences that are greater than the difference in the original sample means.
If the alternative hypothesis is two-tailed, the \(p\)-value equals the proportion of the differences that are farther away from 0 than is the difference in the original sample means.
Let’s say we have the sample 17, 20, 22, 23, 15, 18, 24, 25, 18 from one population and the sample 27, 20, 28, 25, 25, 27, 23 from another population.
x = c(17, 20, 22, 23, 15, 18, 24, 25, 18)
n1 = length(x)
mean.x = mean(x)
y = c(27, 20, 28, 25, 25, 27, 23)
n2 = length(y)
mean.y = mean(y)
pool = c(x, y)
n = length(pool)
R = 5000
diff = c()
for (i in 1:R){
indices = sample(1:n, size = n1, replace = FALSE) # Sample indices
xx = pool[indices]
yy = pool[-indices]
diff[i] = mean(xx) - mean(yy)
}
# Another way to do the for loop
for (i in 1:R){
indices = sample(1:n, replace = FALSE) # Permute all indices
xx = pool[head(indices,n1)]
yy = pool[tail(indices,n2)]
diff[i] = mean(xx) - mean(yy)
}The randomization distribution is plotted below:
hist(diff, main = "The Randomization Distribution for the Difference between Sample Means", xlab = "Mean Differences")
abline(v = mean.x-mean.y, lty = "dashed", col = "red")
legend("topright", legend = c("Difference in Sample Means"), lty = 2, col = c("red"), text.col = c("red"), bty = "n")
If we want to test \(H_0: \mu_1=\mu_2\) vs. \(H_a: \mu_1 < \mu_2\), the \(p\)-value would be equal to the proportion of differences less than the difference in original sample means:
(sum(diff < mean.x-mean.y))/R## [1] 0.005and the \(p\)-value based on the traditional two-sample t test is
t.test(x, y, alternative = "less")##
## Welch Two Sample t-test
##
## data: x and y
## t = -3.0698, df = 13.968, p-value = 0.004168
## alternative hypothesis: true difference in means is less than 0
## 95 percent confidence interval:
## -Inf -2.036026
## sample estimates:
## mean of x mean of y
## 20.22222 25.00000The two \(p\)-values appear to be similar.
Bootstrapping Confidence Interval for Correlation
The eye widths (in cm) and eyelash lengths (in cm) for a sample of 22 mammals are given below:
D = data.frame(width=c("Eye width", 0.57, 0.39, 0.74, 0.58, 1.08, 0.72, 0.70, 1.52, 0.92, 1.75, 1.89, 1.98, 1.98, 2.02, 2.20, 2.41, 2.54, 2.60, 2.64, 2.92, 3.09, 4.08),
length = c("Eyelash length", 0.13, 0.23, 0.23, 0.41, 0.43, 0.24, 0.36, 0.32, 0.61, 0.42, 0.60, 0.38, 0.47, 0.95, 0.76, 0.89, 0.86, 1.21, 0.76, 0.80, 0.67, 1.66))
names(D) = NULL
kable(D, align='cc',
caption = "<center><strong>The Eye Width and Eyelash Length of 22 Mammals</strong></center>",
table.attr = "style='width:60%; '") %>%
footnote("From Statististics: Concepts & Controversies by David S. Moore and William I. Notz ")%>%
kable_classic(full_width = TRUE, position = "center") %>%
column_spec (1:2,border_left = TRUE, border_right = TRUE) %>%
kable_styling("bordered") # Options: basic, striped, bordered, hover, condensed, responsive and none.| Eye width | Eyelash length |
| 0.57 | 0.13 |
| 0.39 | 0.23 |
| 0.74 | 0.23 |
| 0.58 | 0.41 |
| 1.08 | 0.43 |
| 0.72 | 0.24 |
| 0.7 | 0.36 |
| 1.52 | 0.32 |
| 0.92 | 0.61 |
| 1.75 | 0.42 |
| 1.89 | 0.6 |
| 1.98 | 0.38 |
| 1.98 | 0.47 |
| 2.02 | 0.95 |
| 2.2 | 0.76 |
| 2.41 | 0.89 |
| 2.54 | 0.86 |
| 2.6 | 1.21 |
| 2.64 | 0.76 |
| 2.92 | 0.8 |
| 3.09 | 0.67 |
| 4.08 | 1.66 |
| Note: | |
| From Statististics: Concepts & Controversies by David S. Moore and William I. Notz |
Let’s look at the scatterplot:
myData = data.frame(width=c(0.57, 0.39, 0.74, 0.58, 1.08, 0.72, 0.70, 1.52, 0.92, 1.75, 1.89, 1.98, 1.98, 2.02, 2.20, 2.41, 2.54, 2.60, 2.64, 2.92, 3.09, 4.08),
length = c( 0.13, 0.23, 0.23, 0.41, 0.43, 0.24, 0.36, 0.32, 0.61, 0.42, 0.60, 0.38, 0.47, 0.95, 0.76, 0.89, 0.86, 1.21, 0.76, 0.80, 0.67, 1.66))
plot(length~width, data = myData)
The plot suggests a strong correlation between length and width.
The following uses the “boot” package to find a confidence interval for the population correlation \(\rho\).
# Step 1: define the R function that calculates each bootstrap statistic
correlation = function(d, i){
D = d[i, ]
cor(D$width, D$length)
}
# Step 2: Do bootstrapping and call the function to calculate bootstrap statistics
b = boot(myData, correlation, R = 5000)
# The following shows the bootstrap distribution
hist(b$t, main = "Histogram of Bootstrap Correlations", xlab = "Bootstrap correlation")
abline(v = b$t0, lty = "dashed", col = "red")
legend("topleft", legend=c('Observed Correlation'), lty="dashed", col=c('red'), text.col = c('red'), bty = "n")
# Step 3: Calculate 95% confidence interval bases on
# the bootstrap percentile or bca method
boot.ci(b, conf = 0.95, type = c("perc", "bca"))## BOOTSTRAP CONFIDENCE INTERVAL CALCULATIONS
## Based on 5000 bootstrap replicates
##
## CALL :
## boot.ci(boot.out = b, conf = 0.95, type = c("perc", "bca"))
##
## Intervals :
## Level Percentile BCa
## 95% ( 0.6867, 0.9339 ) ( 0.6893, 0.9358 )
## Calculations and Intervals on Original ScaleWe can use the parametric method to find a 95% confidence interval for the population correlation.
cor.test(myData$width, myData$length, method = "pearson", conf.level = 0.95)##
## Pearson's product-moment correlation
##
## data: myData$width and myData$length
## t = 7.0267, df = 20, p-value = 8.137e-07
## alternative hypothesis: true correlation is not equal to 0
## 95 percent confidence interval:
## 0.6549864 0.9332853
## sample estimates:
## cor
## 0.8436286Obtaining P-value for the Test about Correlation with Resampling
The following uses the randomization method to find the \(p\)-value for testing \(\rho=0\) vs. \(\rho \ne 0\). To calculate the \(p\)-value, we need to assume that the null hypothesis is true. Under the null hypothesis, the order of width values does not matter, so we permute the width column each time we repeat the randomization.
Note: When we bootstrap for correlations, we keep all width and length pairs together, and randomly sample pairs with replacement. But in randomization, we permute one column and holding the other column fixed. The bootstrap distribution should center around the observed sample correlation, whereas the randomization distribution should center around 0.
We first plot the correlations based on all randomizations.
R = 5000
n = nrow(myData)
r = c()
# Calculate bootstrap statistics under the null hypothesis that there is correlation
for (i in 1:R) {
s <- sample(myData$width, replace = FALSE) # Shuffle the width column, while keeping length column
r[i] <- cor(s, myData$length)
}
# Plot the randomization distribution
hist(r, main = "Randomization Distribution of r", breaks = 30 , xlim = c(-1,1))
# Calculate the observed statistic (i.e., the sample correlation)
observed.r = cor(myData$width, myData$length)
abline(v = observed.r, lty = "dashed", col = "red")
legend("topleft", legend=c('Observed Correlation'), lty="dashed", col=c('red'), text.col = c('red'), bty = "n")
We then calculate the \(p\)-value:
# The p-value is the total area at the two tails, one to the right of the
# sample correlation and one to the left of negative sample correlation.
sum(r >= observed.r | r <= -observed.r)/R## [1] 0# Or by
sum(abs(r-0)>=abs(observed.r-0))/R## [1] 0We can directly test \(H_0: \rho=0\) vs \(H_a: \rho \ne 0\):
cor.test(myData$width, myData$length, method = "pearson", alternative = c("two.sided"))##
## Pearson's product-moment correlation
##
## data: myData$width and myData$length
## t = 7.0267, df = 20, p-value = 8.137e-07
## alternative hypothesis: true correlation is not equal to 0
## 95 percent confidence interval:
## 0.6549864 0.9332853
## sample estimates:
## cor
## 0.8436286The randomization method and the traditional method give similar results.
Chapter 3. Sign Test
A reference is here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC153434/
A signed test is used in two situations:
for testing a single population median or mean (only when the population distribution is symmetric) based on one sample
for testing the difference in two means based on two samples that are paired.
When dealing with paired samples, always work on the differences for each pair so that the data essentially become one sample.
Sign Test for One-Sample Data
Let \(m\) denote the median of a population. The following are steps to run the sign test.
Step 1. Choose the hypotheses. The null hypothesis is \(H_0\): \(m = m_0\). The alternative hypothesis is one of the following:
\(H_a\): \(m < m_0\),
\(H_a\): \(m > m_0\), or
\(H_a\): \(m \ne m_0\).
Step 2. Replace each value in the data with a “+” sign, a “-” sign, or an “NA”, depending on whether the value is greater than, less than, or equal to \(m_0\). Let \(n\) denote the number of “-” or “-” signs, \(N_+\) denote the number of “+” signs, \(N_-\) denote the number of “-” signs, and \(S\) denote the smaller of \(N_+\) and \(N_-\).
Step 3. The test statistic is \(N_+\). Under the null hypothesis \(H_0\): \(m = m_0\), \(N_+\) has a binomial distribution with success probability \(p = 0.5\), so
the left-sided alternative \(H_a: m < m_0\) becomes \(H_a: p < 0.5\),
the right-sided alternative \(H_a: m > m_0\) becomes \(H_a: p > 0.5\), and
the two-sided alternative \(H_a: m \ne m_0\) becomes \(H_a: p \ne 0.5\),
Step 4. The \(p\)-value is the probability of obtaining test results at least as extreme as the result actually observed, under the assumption that the null hypothesis is true. Specifically,
for the left-sided alternative \(H_a: p < 0.5\), the \(p\)-value equals \(P(N_+<x)\), where \(x\) represents the observed number of “+” signs.
for the right-sided alternative \(H_a: p > 0.5\), the \(p\)-value equals \(P(N_+>x)\), where \(x\) represents the observed number of “+” signs.
for the two-sided alternative \(H_a: p \ne 0.5\), the \(p\)-value equals twice the smaller of the previous two values.
Step 5. Make a decision and draw a conclusion in the context of the problem.
Equally, we can use the following R code for calculating the p-value of a sign test:
\[binom.test(x, n, \text{alternative = c("two.sided", "less", "greater")})\] where \(x\) is the number of “+” signs and \(n\) is the number of “+” or “-” signs.
Example 1.
A bank manager claims that the mean number of customer per day is less than 750. A teller doubts the accuracy of this claim. The number of bank customers per day for 16 randomly selected days are listed below. At 0.05 significance level, can the teller reject the bank manager’s claim?
\[775, 765, 801, 742, 754, 753, 739, 751, 745, 750, 777, 769, 756, 760, 782, 789\]
Use the one-sample t-test and the sign test to test the claim that the mean number of customer per day is less than 750. Choose the significance level \(\alpha = 0.05\).
Solution.
Since the test is about the population mean, to use the sign test, we must assume that the population distribution is symmetric, though it does not have to be normal.
Step 1. \(H_0\): \(m = 750\) vs \(H_0\): \(m < 750\)
Step 2. The signs are + + + - + + - + - NA + + + + + +
Step 3. The value of the test statistic is 12.
Step 4. The \(p\)-value equals \(P(N_+\le 12)=0.9963\), where \(N_+\) has a binomial distribution with \(p=0.5\). Here I used the R code “pbinom(12, 15, 0.5)” for calculating \(P(N_+\le 12)\), where the number 15 is the number of “+” or “-” signs.
Step 5. Since the \(p\)-value (0.9963) is greater than \(\alpha\), we do not reject the null hypothesis. We conclude that the data do not provide sufficient evidence that the mean number of customer per day is less than 750.
The \(p\)-value can also be calculated using the following code:
binom.test(12, 15, alternative = "less")##
## Exact binomial test
##
## data: 12 and 15
## number of successes = 12, number of trials = 15, p-value = 0.9963
## alternative hypothesis: true probability of success is less than 0.5
## 95 percent confidence interval:
## 0.0000000 0.9431531
## sample estimates:
## probability of success
## 0.8If we use the ordinary one-sample t test, we need to assume that the population has a normal distribution. The R code for the \(p\)-value of this test is
x = c(775, 765, 801, 742, 754, 753, 739, 751, 745, 750, 777, 769, 756, 760, 782, 789)
t.test(x, mu = 750, alternative = "less")##
## One Sample t-test
##
## data: x
## t = 2.9209, df = 15, p-value = 0.9947
## alternative hypothesis: true mean is less than 750
## 95 percent confidence interval:
## -Inf 770.8022
## sample estimates:
## mean of x
## 763The \(p\)-value based on the one-sample t test with a left-sided alternative is 0.9947. Since this \(p\)-value is greater than \(\alpha\), we do not reject the null hypothesis. We conclude that the data do not provide sufficient evidence that the mean number of customer per day is less than 750.
The \(p\)-value is similar to the one obtained with the sign test.
Example 2.
The following are measures of the breaking strength of a certain kind of 2-inch cotton ribbon in pounds:
163, 165, 160, 189, 161, 171, 158, 151, 169, 162, 163, 139, 172, 165, 148, 166, 172, 163, 187, 173
Run the one-sample t-test and the sign test to test that the mean breaking strength is greater than 160. Assume the population distribution of breaking strength is symmetric.
Solution.
Step 1. The null hypothesis is \(H_0: \mu = 160\) and the alternative hypothesis is \(H_a: \mu > 160\).
Step 2. Count the positive differences between the observed vales and the hypothesized value 160.
## + + NA + + + - - + + + - + + - + + + + +Step 3. The value of the test statistic is 15, since 15 out of 19 non-zeros are plus signs.
Step 4. Determine the \(p\)-value using a binomial distribution.
Since 15 out of 19 are plus signs, the \(p\)-value equals \(P(N_+\ge15)\), where \(N_+\) has a binomial distribution with \(n=19\) and \(p=0.5\). Using the following R code
\[1-pbinom(14, 19, 0.5)\] we obtain the \(p\)-value 0.0096.
Step 5. Make a decision and draw a conclusion.
Since the \(p\)-value (0.0096) is less than \(\alpha\), we do reject the null hypothesis. We conclude that the data provide sufficient evidence that the mean breaking strength is greater than 160.
Using the following code, we can directly get the \(p\)-value:
binom.test(15, 19, alternative = "greater")##
## Exact binomial test
##
## data: 15 and 19
## number of successes = 15, number of trials = 19, p-value = 0.009605
## alternative hypothesis: true probability of success is greater than 0.5
## 95 percent confidence interval:
## 0.5808798 1.0000000
## sample estimates:
## probability of success
## 0.7894737Example 3.
Suppose you manage a zoo and you hypothesize that the mean age of children visiting is 8. This becomes your null hypothesis, \(H_0\): the median age of children visiting is 8. The alternative hypothesis is \(H_a\): the median age of children visiting is NOT 8.
Suppose now you took a sample of ten children whose ages turn out to be
\[10, 12, 2, 4, 5, 17, 1, 2, 15, 7\]
Use the one-sample t-test and the sign test to test the claim that the mean age of children visiting is NOT 8.
Solution.
Step 1. \(H_0\): the median age of children visiting is 8. The alternative hypothesis is \(H_a\): the median age of children visiting is NOT 8.
Step 2. The signs of data (“+” when > 8 and “-” when < 8) are: +, +, -, -, -, +, -, -, +, -.
Step 3. The value of the test statistic is 4.
Step 4. The \(p\)-value equals \(2*P(N_+\le 4)=2*0.37695 = 0.7539\), where \(N_+\) has a binomial distribution with \(p=0.5\). Here I used the R code “pbinom(4, 10, 0.5)” for calculating \(P(N_+\le 4)\), where the number 10 is the number of “+” or “-” signs.
Step 5. Since the \(p\)-value (0.7539) is greater than \(\alpha\), we do not reject the null hypothesis. We conclude that the data do not provide sufficient evidence that the mean age of children visiting is NOT 8.
The \(p\)-value can also be calculated using the following code:
binom.test(4, 10, alternative = "two.sided")##
## Exact binomial test
##
## data: 4 and 10
## number of successes = 4, number of trials = 10, p-value = 0.7539
## alternative hypothesis: true probability of success is not equal to 0.5
## 95 percent confidence interval:
## 0.1215523 0.7376219
## sample estimates:
## probability of success
## 0.4If we use the ordinary one-sample t test, we need to assume that the population has a normal distribution. The R code for the \(p\)-value of this test is
x = c(10, 12, 2, 4, 5, 17, 1, 2, 15, 7)
t.test(x, mu = 8, alternative = "two.sided")##
## One Sample t-test
##
## data: x
## t = -0.27641, df = 9, p-value = 0.7885
## alternative hypothesis: true mean is not equal to 8
## 95 percent confidence interval:
## 3.407919 11.592081
## sample estimates:
## mean of x
## 7.5The \(p\)-value based on the one-sample t test with a two-sided alternative is 0.7885. Since this \(p\)-value is greater than \(\alpha\), we do not reject the null hypothesis. We conclude that the data do not provide sufficient evidence that the mean number of customer per day is less than 750.
The \(p\)-value is similar to the one obtained with the sign test.
Sign Test for Two-Sample Paired Data
The sign test can also be used when dealing with paired data. In such problems, each pair of sample values is replaced by a “+” sign if the first value exceeds the second value, by a “-” sign if the first value is less than the second, and by an “NA” if the first value equals the second. To test the null hypothesis that two continuous populations have equal medians (or symmetrical populations have equal means), we can thus use the sign test and call it the paired-sample sign test.
We can just apply the one-sample sign test to the differences.
Example 4.
A psychologist claims that the mean number of repeat offenders will decrease if first time offenders complete a particular rehabilitation course. You randomly select 10 prisons and record the number of repeat offenders during a two-year period. Then, after first-time offenders complete the course, you record the number of repeat offenders at each prison for another two-year period. The results are shown in the following table. At 0.05 significance level, can you support the psychologist’s claim?
| prision | before | after |
| 1 | 21 | 19 |
| 2 | 34 | 22 |
| 3 | 9 | 16 |
| 4 | 45 | 31 |
| 5 | 30 | 21 |
| 6 | 54 | 30 |
| 7 | 37 | 22 |
| 8 | 36 | 18 |
| 9 | 33 | 17 |
| 10 | 40 | 21 |
| Note: | ||
Use the paired-sample t-test and the paired-sample sign test to test the claim that the mean number of repeat offenders will decrease if first time offenders complete a particular rehabilitation course.
Solution.
Step 1. \(H_0: \mu_1 = \mu_2\) vs. \(H_a: \mu_1 > \mu_2\)
Step 2. The signs (Before - After): + + - + + + + + + +
Step 3. The value of the test statistic is 9.
Step 4. The \(p\)-value equals \(P(N_+\ge 9)=1-P(N_+<9) = 1-P(N_+\le 8) = 1-0.9893 = 0.0107\), where \(N_+\) has a binomial distribution with \(p=0.5\). Here I used the R code “pbinom(8, 10, 0.5)” for calculating \(P(N_+\le 8)\), where the number 10 is the number of “+” or “-” signs.
Step 5. Since the \(p\)-value (0.0107) is greater than \(\alpha\), we reject the null hypothesis. We conclude that the data do provide sufficient evidence that …
If we use the ordinary one-sample t test, we need to assume that the two populations have normal distributions. The R code for the \(p\)-value of this test is
Example 5.
The following data, in tons, are the amounts of sulfur oxides emitted by a large industrial plant in 40 days:
17, 15, 20, 29, 19, 18, 22, 25, 27, 9, 24, 20, 17, 6, 24, 14, 15, 23, 24, 26, 19, 23, 28, 19, 16, 22, 24, 17, 20, 13, 19, 10, 23, 18, 31, 13, 20, 17, 24, 14
Use the one-sample t-test and the sign test to test the claim that the mean amount of sulfur oxides emitted by the plant is less than 21.5.
Solution.
\(H_0: m = 21.5\) vs. \(H_a: m < 21.5\)
The p-value can be obtained by
binom.test(x=16, n=40, p=0.5, alternative="less")##
## Exact binomial test
##
## data: 16 and 40
## number of successes = 16, number of trials = 40, p-value = 0.1341
## alternative hypothesis: true probability of success is less than 0.5
## 95 percent confidence interval:
## 0.0000000 0.5422167
## sample estimates:
## probability of success
## 0.4# or
pbinom(16, 40, 0.5)## [1] 0.1340936Example 6.
In a clinical trial, survival time (weeks) is collected for 10 subjects with non-Hodgkin’s lymphoma. The exact survival time was not known for one subject who was still alive after 362 weeks, when the study ended. The subjects’ survival times were
49, 58, 75, 110, 112, 132, 151, 276, 281, 362+
The plus sign indicates the subject still alive at the end of the study. The researcher wished to determine if the median survival time was less than or greater than 200 weeks.
Use the sign test to test the claim that the median survival time was less than or greater than 200 weeks.
Chapter 4. Wilcoxon Signed-Rank Test
The sign test has an obvious disadvantage that it simply allocates a sign to each observation, according to whether it is less than or greater than some hypothesized value, and does not take the magnitude of the observation into account. Omitting information on the magnitude of the observations is rather inefficient and may reduce the statistical power of the test. The Wilcoxon signed-rank test is an alternative that does account for the magnitude of the observations.
Wilcoxon signed-rank test for One-Sample Data
This Wilcoxon signed-rank test consists of five basic steps:
Step 1. State the null and alternative hypotheses.
Step 2. Determine differences, ranks, and signs. Subtract the hypothesized value from each observation and rank all the differences in increasing order of magnitude (without regard to their sign). Ignore all observations that are equal to the hypothesized value. If two observations have the same magnitude, regardless of sign, then they are given an average ranking. Allocate a sign (+ or -) to each observation according to whether it is greater or less than the hypothesized value (as in the sign test).
Step 3. Calculate the value of the test statistic \(R^+\), the sum of all positive ranks.
Step 4. Calculate the \(p\)-value:
Step 5. Make a decision and draw a conclusion.
Remark: the sum of all non-zero ranks is \(\frac{n(n+1)}{2}\), where \(n\) is the number of non-zero differences. Why?
A great reference: https://online.stat.psu.edu/stat415/lesson/20/20.2
The following shows the details how the \(p\)-value can be calculated.
wilcoxTest = function(n, V, alternative = c("two.sided", "less", "greater"), plot = FALSE){
# Attention: n is the number of non-zero differences
alternative = alternative[1]
W = n*(n+1)/2
x=c(-1,1)
L = lapply(1:n, function(j) x)
D = expand.grid(L)
M = matrix(0, n, n)
diag(M)[1:n]=1:n
D1 = (M%*%as.matrix(t(D)))[1:n, ]
s = apply(D1, 2, function(r) ifelse(all(r<0), 0, sum(r[r>0])))
D2 = rbind(D1, s)
names(D2)=NULL
p0 = sum((table(D2[n+1,])/2^n)[1:(V+1)]) # p-value for left-sided
p1 = sum((table(D2[n+1,])/2^n)[(V+1):(W+1)]) # p-value for right-sided
if(alternative == "less") {
p = p0 # p-value for left-sided
} else if(alternative == "greater") {
p = p1 # p-value for right-sided
} else{
p = 2 * min(p0, p1) # p-value for two-sided alternative
}
L = list(p=p, distribution = table(D2[n+1,])/2^n)
if(plot){
plot(L$distribution, type = "h", ylab = "Probability", xlab = "x (Sum of ranks of positive differences)")
abline(v = V, lty = "dashed", col = "blue")
if (alternative == "less"){
arrows(V, max(L$distribution)/2, 0, col = "red")
} else if(alternative == "greater") {
arrows(V, max(L$distribution)/2, W, col = "red")
} else{
arrows(min(V, W-V), max(L$distribution)/2, 0, , max(L$distribution)/2, col = "red")
arrows(max(V, W-V), max(L$distribution)/2, W, , max(L$distribution)/2, col = "red")
}
}
invisible(L) # Return L but will not be seen directly
}
# Apply the function
wilcoxTest(10, 6, "less")
wilcoxTest(10, 6, "greater")
wilcoxTest(10, 35, "two.sided")We can use the above function to make a useful table similar to one here: https://online.stat.psu.edu/stat415/lesson/20/20.2
| \(n\) | \(R\) | \(x\) | \(R-x\) | \(P(W\le x)=P(W\ge R - x)\) |
| 5 | 15 | 0 | 15 | 0.0312 |
| 1 | 14 | 0.0625 | ||
| 2 | 13 | 0.0938 | ||
| 3 | 12 | 0.1562 | ||
| 4 | 11 | 0.2188 | ||
| 5 | 10 | 0.3125 | ||
| 6 | 9 | 0.4062 | ||
| 7 | 8 | 0.5 | ||
| 6 | 21 | 0 | 21 | 0.0156 |
| 1 | 20 | 0.0312 | ||
| 2 | 19 | 0.0469 | ||
| 3 | 18 | 0.0781 | ||
| 4 | 17 | 0.1094 | ||
| 5 | 16 | 0.1562 | ||
| 6 | 15 | 0.2188 | ||
| 7 | 14 | 0.2812 | ||
| 8 | 13 | 0.3438 | ||
| 9 | 12 | 0.4219 | ||
| 10 | 11 | 0.5 | ||
| 7 | 28 | 0 | 28 | 0.0078 |
| 1 | 27 | 0.0156 | ||
| 2 | 26 | 0.0234 | ||
| 3 | 25 | 0.0391 | ||
| 4 | 24 | 0.0547 | ||
| 5 | 23 | 0.0781 | ||
| 6 | 22 | 0.1094 | ||
| 7 | 21 | 0.1484 | ||
| 8 | 20 | 0.1875 | ||
| 9 | 19 | 0.2344 | ||
| 10 | 18 | 0.2891 | ||
| 11 | 17 | 0.3438 | ||
| 12 | 16 | 0.4062 | ||
| 13 | 15 | 0.4688 | ||
| 14 | 14 | 0.5312 | ||
| 8 | 36 | 0 | 36 | 0.0039 |
| 1 | 35 | 0.0078 | ||
| 2 | 34 | 0.0117 | ||
| 3 | 33 | 0.0195 | ||
| 4 | 32 | 0.0273 | ||
| 5 | 31 | 0.0391 | ||
| 6 | 30 | 0.0547 | ||
| 7 | 29 | 0.0742 | ||
| 8 | 28 | 0.0977 | ||
| 9 | 27 | 0.125 | ||
| 10 | 26 | 0.1562 | ||
| 11 | 25 | 0.1914 | ||
| 12 | 24 | 0.2305 | ||
| 13 | 23 | 0.2734 | ||
| 14 | 22 | 0.3203 | ||
| 15 | 21 | 0.3711 | ||
| 16 | 20 | 0.4219 | ||
| 17 | 19 | 0.4727 | ||
| 18 | 18 | 0.5273 | ||
| 9 | 45 | 0 | 45 | 0.002 |
| 1 | 44 | 0.0039 | ||
| 2 | 43 | 0.0059 | ||
| 3 | 42 | 0.0098 | ||
| 4 | 41 | 0.0137 | ||
| 5 | 40 | 0.0195 | ||
| 6 | 39 | 0.0273 | ||
| 7 | 38 | 0.0371 | ||
| 8 | 37 | 0.0488 | ||
| 9 | 36 | 0.0645 | ||
| 10 | 35 | 0.082 | ||
| 11 | 34 | 0.1016 | ||
| 12 | 33 | 0.125 | ||
| 13 | 32 | 0.1504 | ||
| 14 | 31 | 0.1797 | ||
| 15 | 30 | 0.2129 | ||
| 16 | 29 | 0.248 | ||
| 17 | 28 | 0.2852 | ||
| 18 | 27 | 0.3262 | ||
| 19 | 26 | 0.3672 | ||
| 20 | 25 | 0.4102 | ||
| 21 | 24 | 0.4551 | ||
| 22 | 23 | 0.5 | ||
| 10 | 55 | 0 | 55 | 0.001 |
| 1 | 54 | 0.002 | ||
| 2 | 53 | 0.0029 | ||
| 3 | 52 | 0.0049 | ||
| 4 | 51 | 0.0068 | ||
| 5 | 50 | 0.0098 | ||
| 6 | 49 | 0.0137 | ||
| 7 | 48 | 0.0186 | ||
| 8 | 47 | 0.0244 | ||
| 9 | 46 | 0.0322 | ||
| 10 | 45 | 0.042 | ||
| 11 | 44 | 0.0527 | ||
| 12 | 43 | 0.0654 | ||
| 13 | 42 | 0.0801 | ||
| 14 | 41 | 0.0967 | ||
| 15 | 40 | 0.1162 | ||
| 16 | 39 | 0.1377 | ||
| 17 | 38 | 0.1611 | ||
| 18 | 37 | 0.1875 | ||
| 19 | 36 | 0.2158 | ||
| 20 | 35 | 0.2461 | ||
| 21 | 34 | 0.2783 | ||
| 22 | 33 | 0.3125 | ||
| 23 | 32 | 0.3477 | ||
| 24 | 31 | 0.3848 | ||
| 25 | 30 | 0.4229 | ||
| 26 | 29 | 0.4609 | ||
| 27 | 28 | 0.5 | ||
| 11 | 66 | 0 | 66 | 5e-04 |
| 1 | 65 | 0.001 | ||
| 2 | 64 | 0.0015 | ||
| 3 | 63 | 0.0024 | ||
| 4 | 62 | 0.0034 | ||
| 5 | 61 | 0.0049 | ||
| 6 | 60 | 0.0068 | ||
| 7 | 59 | 0.0093 | ||
| 8 | 58 | 0.0122 | ||
| 9 | 57 | 0.0161 | ||
| 10 | 56 | 0.021 | ||
| 11 | 55 | 0.0269 | ||
| 12 | 54 | 0.0337 | ||
| 13 | 53 | 0.0415 | ||
| 14 | 52 | 0.0508 | ||
| 15 | 51 | 0.0615 | ||
| 16 | 50 | 0.0737 | ||
| 17 | 49 | 0.0874 | ||
| 18 | 48 | 0.103 | ||
| 19 | 47 | 0.1201 | ||
| 20 | 46 | 0.1392 | ||
| 21 | 45 | 0.1602 | ||
| 22 | 44 | 0.1826 | ||
| 23 | 43 | 0.2065 | ||
| 24 | 42 | 0.2324 | ||
| 25 | 41 | 0.2598 | ||
| 26 | 40 | 0.2886 | ||
| 27 | 39 | 0.3188 | ||
| 28 | 38 | 0.3501 | ||
| 29 | 37 | 0.3823 | ||
| 30 | 36 | 0.4155 | ||
| 31 | 35 | 0.4492 | ||
| 32 | 34 | 0.4829 | ||
| 33 | 33 | 0.5171 | ||
| 12 | 78 | 0 | 78 | 2e-04 |
| 1 | 77 | 5e-04 | ||
| 2 | 76 | 7e-04 | ||
| 3 | 75 | 0.0012 | ||
| 4 | 74 | 0.0017 | ||
| 5 | 73 | 0.0024 | ||
| 6 | 72 | 0.0034 | ||
| 7 | 71 | 0.0046 | ||
| 8 | 70 | 0.0061 | ||
| 9 | 69 | 0.0081 | ||
| 10 | 68 | 0.0105 | ||
| 11 | 67 | 0.0134 | ||
| 12 | 66 | 0.0171 | ||
| 13 | 65 | 0.0212 | ||
| 14 | 64 | 0.0261 | ||
| 15 | 63 | 0.032 | ||
| 16 | 62 | 0.0386 | ||
| 17 | 61 | 0.0461 | ||
| 18 | 60 | 0.0549 | ||
| 19 | 59 | 0.0647 | ||
| 20 | 58 | 0.0757 | ||
| 21 | 57 | 0.0881 | ||
| 22 | 56 | 0.1018 | ||
| 23 | 55 | 0.1167 | ||
| 24 | 54 | 0.1331 | ||
| 25 | 53 | 0.1506 | ||
| 26 | 52 | 0.1697 | ||
| 27 | 51 | 0.1902 | ||
| 28 | 50 | 0.2119 | ||
| 29 | 49 | 0.2349 | ||
| 30 | 48 | 0.2593 | ||
| 31 | 47 | 0.2847 | ||
| 32 | 46 | 0.311 | ||
| 33 | 45 | 0.3386 | ||
| 34 | 44 | 0.3667 | ||
| 35 | 43 | 0.3955 | ||
| 36 | 42 | 0.425 | ||
| 37 | 41 | 0.4548 | ||
| 38 | 40 | 0.4849 | ||
| 39 | 39 | 0.5151 |
Example.
The following are 15 measurements of the octane rating of a certain kind of gasoline:
97.5, 95.2, 97.3, 96, 96.8, 100.3, 97.4, 95.3, 93.2, 99.1, 96.1, 97.6, 98.2, 98.5, 94.9
Use the traditional paired-sample t test and the Wilcoxon signed-rank test to test whether the mean octane rating of the given kind of gasoline is 98.5.
Solution.
Step 1. \(H_0: \mu = 98.5\) vs. \(H_a: \mu \ne 98.5\)
Step 2. Add differences, ranks, and signs.
| Rating | Diff | Rank | Sign |
| 97.5 | -1 | 4 | − |
| 95.2 | -3.3 | 12 | − |
| 97.3 | -1.2 | 6 | − |
| 96 | -2.5 | 10 | − |
| 96.8 | -1.7 | 7 | − |
| 100.3 | 1.8 | 8 | + |
| 97.4 | -1.1 | 5 | − |
| 95.3 | -3.2 | 11 | − |
| 93.2 | -5.3 | 14 | − |
| 99.1 | 0.6 | 2 | + |
| 96.1 | -2.4 | 9 | − |
| 97.6 | -0.9 | 3 | − |
| 98.2 | -0.3 | 1 | − |
| 98.5 | 0 | 0 | NA |
| 94.9 | -3.6 | 13 | − |
Step 3. The value of the test statistic (the sum of ranks of positive differences: \(R^+\)) is
sum(D$Rank * (D$Diff >0))## [1] 10Step 4. Determine the \(p\)-value. Using the previous table with \(n=14\) (a value that equals the hypothesized value was removed) and test statistic value 10, we get the \(p\)-value \((0.0026)\cdot 2=0.0052\).
Step 5. Since the \(p\)-value (0.0052) is less than the significance level (\(\alpha=0.05\)), we reject the null hypothesis and conclude that the data provide sufficient evidence that the mean octane rating of the given kind of gasoline is NOT 98.5.
Note: We can use the “wilcox.test()” function to carry out the Wilcoxon signed-rank test:
# We first remove the observation with rating equal to the hypothesized value
D = D[D$Diff != 0,]
wilcox.test(D$Rating, mu = 98.5, alternative = "two.sided", exact = TRUE)##
## Wilcoxon signed rank exact test
##
## data: D$Rating
## V = 10, p-value = 0.005249
## alternative hypothesis: true location is not equal to 98.5The \(p\)-value is the same as we obtained by using the table.
Exercise.
The length (in cm) of 10 randomly selected pygmy sunfish are
5.0, 3.9, 5.2, 5.5, 2.8, 6.1, 6.4, 2.6, 1.7, 4.3
Can we conclude that the median length of pygmy sunfish differs significantly from 3.7 centimeters?
Wilcoxon signed-rank test for Paired Data
The Wilcoxon signed-rank test can also be used for paired data. Here is an example.
Example.
The following are the weights in pounds, before and after, of 16 people who stayed on a certain reducing diet for four weeks:
D = data.frame(Before = c(147.0, 183.5, 232.1, 161.6, 197.5, 206.3, 177.0, 215.4, 147.7, 208.1, 166.8, 131.9, 150.3, 197.2, 159.8, 171.7),
After = c(137.9, 176.2, 219.0, 163.8, 193.5, 201.4, 180.6, 203.2, 149.0, 195.4, 158.5, 134.4, 149.3, 189.1, 159.1, 173.2)
)
D1 = rbind(c("Before", "After"), D)
names(D1) = NULL
kable(D1, align='cc',
caption = "<center><strong>Paired Data</strong></center>",
table.attr = "style='width:80%; '") %>%
kable_classic(full_width = TRUE, position = "center") %>%
column_spec (1:2,border_left = TRUE, border_right = TRUE) %>%
kable_styling("bordered") # Options: basic, striped, bordered, hover, condensed, responsive and none.| Before | After |
| 147 | 137.9 |
| 183.5 | 176.2 |
| 232.1 | 219 |
| 161.6 | 163.8 |
| 197.5 | 193.5 |
| 206.3 | 201.4 |
| 177 | 180.6 |
| 215.4 | 203.2 |
| 147.7 | 149 |
| 208.1 | 195.4 |
| 166.8 | 158.5 |
| 131.9 | 134.4 |
| 150.3 | 149.3 |
| 197.2 | 189.1 |
| 159.8 | 159.1 |
| 171.7 | 173.2 |
Use the traditional paired-sample t test and the Wilcoxon signed-rank test to test whether the weight-reducing diet is effective.
Solution.
Step 1. \(H_0: m_1 = m_2\) vs. \(H_a: m_1 > m_2\)
Step 2. Add differences, ranks, and signs.
D$Diff = round(D$Before - D$After, 1) # Without rounding, many digits are displayed
D$Rank = rank(abs(D$Diff))
D$Sign = ifelse(D$Diff<0, "−", ifelse(D$Diff>0, "\\+", "NA"))
D1 = rbind(c("Before", "After", "Diff", "Rank", "Sign"), D)
names(D1) = NULL
kable(D1, align='ccccc',
caption = "<center><strong>Paired Data with Differences and Ranks</strong></center>",
table.attr = "style='width:80%; '") %>%
kable_classic(full_width = TRUE, position = "center") %>%
column_spec (1:5,border_left = TRUE, border_right = TRUE) %>%
kable_styling("bordered") # Options: basic, striped, bordered, hover, condensed, responsive and none.| Before | After | Diff | Rank | Sign |
| 147 | 137.9 | 9.1 | 13 | + |
| 183.5 | 176.2 | 7.3 | 10 | + |
| 232.1 | 219 | 13.1 | 16 | + |
| 161.6 | 163.8 | -2.2 | 5 | − |
| 197.5 | 193.5 | 4 | 8 | + |
| 206.3 | 201.4 | 4.9 | 9 | + |
| 177 | 180.6 | -3.6 | 7 | − |
| 215.4 | 203.2 | 12.2 | 14 | + |
| 147.7 | 149 | -1.3 | 3 | − |
| 208.1 | 195.4 | 12.7 | 15 | + |
| 166.8 | 158.5 | 8.3 | 12 | + |
| 131.9 | 134.4 | -2.5 | 6 | − |
| 150.3 | 149.3 | 1 | 2 | + |
| 197.2 | 189.1 | 8.1 | 11 | + |
| 159.8 | 159.1 | 0.7 | 1 | + |
| 171.7 | 173.2 | -1.5 | 4 | − |
Step 3. The value of the test statistic \(R^+=111\).
Step 4. Calculate the \(p\)-value.
We can use the “wilcox.test()” function to carry out the Wilcoxon signed-rank test:
x = D$Before
y = D$After
wilcox.test(x, y, paired=TRUE, alternative = "greater", exact = TRUE)##
## Wilcoxon signed rank exact test
##
## data: x and y
## V = 111, p-value = 0.01248
## alternative hypothesis: true location shift is greater than 0Note 1: set paired = TRUE!!!!
Note 2: setting exact = TRUE allows us to get a result that is the same as we would do by hand. If it is set to FALSE, R will use a continuity correction when calculating the \(p\)-value. The two methods should give close results if \(n\) is not too small.
Step 5. Since the \(p\)-value (0.0125) is less than the significance level (\(\alpha=0.05\)), we reject the null hypothesis and conclude that the data provide sufficient evidence that the weight-reducing diet is effective.
Chapter 5. Wilcoxon Rank-Sum Test
The Wilcoxon rank-sum test (or equivalently Mann-Whitney U test) is a nonparametric alternative to the two-sample t-test based on two independent samples. The null hypothesis is that the two population have the same distribution (and thus the median). To find an appropriate test statistic, the two samples are pooled and then ranked in increasing order with the smallest value receives the rank 1 (if two values in the pooled data tie, each receives the average of the ranks that would be assigned to them). The test statistic is chosen to be the sum of the ranks of the values in the first sample and is denoted by \(W_1\).
If the alternative hypothesis is that the median of the first population is smaller than that of the second population, then the smaller the test statistic, the more evidence to reject the null hypothesis.
If the alternative hypothesis is that the median of the first population is larger than that of the second population, then the larger the test statistic, the more evidence to reject the null hypothesis.
If the alternative hypothesis is that the medians are the same, then the smaller or larger the test statistic, the more evidence to reject the null hypothesis.
Usually, the test statistic \(W_1\) is replaced by an equivalent test statistic \(U_1 = W_1 - \frac{n_1 (n_1 +1)}{2}\), where \(n_1\) and \(n_2\) are the sample sizes of the two samples, respectively. The “wilcox.test()” function that does Wilcoxon rank-sum test in R prints the test statistic as \(W\).
Let \(W_2\) denote the sum of the ranks of the values in the second sample and let \(U_2 = W_2 - \frac{n_2 (n_2 +1)}{2}\). It is easy to show that
\(W_1+W_2 = \frac{(n_1+n_2)(n_1+n_2 +1)}{2}\) and \(U_1+U_2 =n_1n_2\).
Under the null hypothesis, the random variables \(U_1\) and \(U_2\) have the same mean \(\frac{n_1 n_2}{2}\), and the same variance \(\frac{n_1 n_2 (n_1+n_2+1)}{12}\).
Remark: Mann and Whitney proposed their equivalent test using \(U_2\) as the test statistic. This statistic is obtained by ordering all \(n_1+n_2\) observations from smallest to largest and counting the number of observations in sample one that precede each observation in sample two. The statistic \(U_2\) is the sum of these counts. For example, if sample one is 23, 15, 9, and 22, and sample two is 8, 34, 25, 21, and 45, then the pooled sample after ordering is 8, (9), (15), 21, (22), (23), 25, 34, and 45, where numbers in parentheses are from sample one. Since 0 value from sample one precedes 8, 2 values from sample one precedes 21, 4 values from sample one precedes 25, 4 values from sample one precedes 34, and 4 values from sample one precedes 45, the sum of these counts \(0+2+4+4+4=14\). On the other hand, \(W_2=1+4+7+8+9=29\) and \(U_2=W_2-\frac{5(5+1)}{2}=29-15=14\).
Example 1.
The following are the weight gains (in pounds) of two random samples of young turkeys fed different diets but otherwise kept under identical conditions:
Diet 1: 16.3, 10.1, 10.7, 13.5, 14.9, 11.8, 14.3, 10.2, 12.0, 14.7, 23.6, 15.1, 14.5, 18.4, 13.2, 14.0
Diet 2: 21.3, 23.8, 15.4, 19.6, 12.0, 13.9, 18.8, 19.2, 15.3, 20.1, 14.8, 18.9, 20.7, 21.1, 15.8, 16.2
Use the two-sample t-test and the Wilcoxon rank-sum test to test the claim that on average, the second diet produces a greater gain in weight.
Solution.
The two-sample t test:
Diet1 = c(16.3, 10.1, 10.7, 13.5, 14.9, 11.8, 14.3, 10.2, 12.0, 14.7, 23.6, 15.1, 14.5, 18.4, 13.2, 14.0)
Diet2 = c(21.3, 23.8, 15.4, 19.6, 12.0, 13.9, 18.8, 19.2, 15.3, 20.1, 14.8, 18.9, 20.7, 21.1, 15.8, 16.2)
# Side-by-side boxplots
boxplot(Diet1,Diet2) # An outlier in sample 1, thus evidence of departure from normality
#Use the Shapiro-Wilk normality test to check normality
shapiro.test(Diet1) # p-value 0.04026, some evidence of departure from normality##
## Shapiro-Wilk normality test
##
## data: Diet1
## W = 0.88102, p-value = 0.04026shapiro.test(Diet2) # p-value 0.7341, no significant evidence of departure from normality##
## Shapiro-Wilk normality test
##
## data: Diet2
## W = 0.96397, p-value = 0.7341t.test(Diet1, Diet2, alternative = "less") # Her "less" indicates pop'n 1 has smaller values##
## Welch Two Sample t-test
##
## data: Diet1 and Diet2
## t = -3.1979, df = 29.953, p-value = 0.00163
## alternative hypothesis: true difference in means is less than 0
## 95 percent confidence interval:
## -Inf -1.747867
## sample estimates:
## mean of x mean of y
## 14.20625 17.93125The small \(p\)-value (0.00163) indicates rejection of the null hypothesis at significance level 0.05. We conclude that the data provide sufficient evidence that on average, the second diet produces a greater gain in weight.
The Wilcoxon rank-sum test:
wilcox.test(Diet1, Diet2, alternative = "less")## Warning in wilcox.test.default(Diet1, Diet2, alternative = "less"): cannot
## compute exact p-value with ties##
## Wilcoxon rank sum test with continuity correction
##
## data: Diet1 and Diet2
## W = 45.5, p-value = 0.0009982
## alternative hypothesis: true location shift is less than 0The small \(p\)-value (0.0009982) indicates rejection of the null hypothesis at significance level 0.05. We conclude that the data provide sufficient evidence that on average, the second diet produces a greater gain in weight.
Note: R gives warning messages when there is any tie from the two samples.
Example 2.
Cancer treatment using chemotherapy employs chemicals that kill both cancer cells and normal cells. In some instances, the toxicity of the cancer drug- that is, its effect on normal cells- can be reduced by the simultaneous injection of a second drug. A study was conducted to determine whether a particular drug injection was beneficial in reducing the harmful effects of a chemotherapy treatment on the survival time for rats. Two randomly selected groups of rats, 12 in each group, were used for the experiment. Both groups, call them A and B, received the toxic drug in a dosage large enough to cause death, but group B also received the antitoxin that was intended to reduce the toxic effect of the chemotherapy on normal cells. The test was terminated at the end of 20 days, or 480 hours. The length of survival time for the two groups of rats, to the nearest 4 hours, are given below.
Only Chemotherapy (A): 84, 128, 168, 92, 184, 92, 76, 104, 72, 180, 144, 120
Chemotherapy plus Drug (B): 140, 184, 368, 96, 480, 188, 480, 244, 440, 380, 480, 196
Do the data, at the significance level 0.05, provide sufficient evidence to indicate that rats receiving the antotoxin tended to survival longer after chemotherapy than those not receiving the antitoxin? Use both the 2-sample t test and the Wilcoxon rank-sum test.
Solution.
The two-sample t test:
A = c(84, 128, 168, 92, 184, 92, 76, 104, 72, 180, 144, 120)
B = c(140, 184, 368, 96, 480, 188, 480, 244, 440, 380, 480, 196)
# Side-by-side boxplots
boxplot(A, B) # no significant evidence of departure from normality
#Use the Shapiro-Wilk normality test to check normality
shapiro.test(A) # p-value 0.1878, not significant evidence of departure from normality##
## Shapiro-Wilk normality test
##
## data: A
## W = 0.90568, p-value = 0.1878shapiro.test(B) # p-value 0.08035, not significant evidence of departure from normality##
## Shapiro-Wilk normality test
##
## data: B
## W = 0.87705, p-value = 0.08035t.test(A, B, alternative = "less")##
## Welch Two Sample t-test
##
## data: A and B
## t = -4.2527, df = 12.679, p-value = 0.0004975
## alternative hypothesis: true difference in means is less than 0
## 95 percent confidence interval:
## -Inf -108.3938
## sample estimates:
## mean of x mean of y
## 120.3333 306.3333The small \(p\)-value (0.0004975) indicates rejection of the null hypothesis at significance level 0.05. We conclude that the data provide sufficient evidence that rats receiving the antotoxin tended to survival longer after chemotherapy than those not receiving the antitoxin.
The Wilcoxon rank-sum test:
wilcox.test(A, B, alternative = "less")## Warning in wilcox.test.default(A, B, alternative = "less"): cannot compute exact
## p-value with ties##
## Wilcoxon rank sum test with continuity correction
##
## data: A and B
## W = 11.5, p-value = 0.0002616
## alternative hypothesis: true location shift is less than 0The small \(p\)-value (0.0002616) indicates rejection of the null hypothesis at significance level 0.05. We conclude that the data provide sufficient evidence that rats receiving the antotoxin tended to survival longer after chemotherapy than those not receiving the antitoxin.
Note: R gives warning messages when there is any tie from the two samples.
Example 3.
Given below are wing stroke frequencies for samples of two species of EUGlossine bees.
Euglossa mandibularis Friese: 235, 225, 190, 188
Euglossa imperialis Cockerell: 180, 169, 180, 185, 178, 183
Do the data, at the significance level 0.05, provide sufficient evidence to indicate that the distributions of wing stroke frequencies for the two species? Use both the 2-sample t test and the Wilcoxon rank-sum test.
Solution.
The two-sample t test:
x = c(235, 225, 190, 188)
y = c(180, 169, 180, 185, 178, 183)
# Side-by-side boxplots
boxplot(x, y) # An outlier in sample 2 indicates its departure from normality
#Use the Shapiro-Wilk normality test to check normality
shapiro.test(x) # p-value 0.1915, not significant evidence of departure from normality##
## Shapiro-Wilk normality test
##
## data: x
## W = 0.83865, p-value = 0.1915shapiro.test(y) # p-value 0.282, not significant evidence of departure from normality##
## Shapiro-Wilk normality test
##
## data: y
## W = 0.88277, p-value = 0.282t.test(x, y, alternative = "two.sided")##
## Welch Two Sample t-test
##
## data: x and y
## t = 2.4802, df = 3.2158, p-value = 0.08357
## alternative hypothesis: true difference in means is not equal to 0
## 95 percent confidence interval:
## -7.151974 67.818641
## sample estimates:
## mean of x mean of y
## 209.5000 179.1667The small \(p\)-value (0.08357) indicates failure to reject the null hypothesis at significance level 0.05. We conclude that the data do not provide sufficient evidence that the distributions of wing stroke frequencies for the two species differ.
The Wilcoxon rank-sum test:
wilcox.test(x, y, alternative = "two.sided")## Warning in wilcox.test.default(x, y, alternative = "two.sided"): cannot compute
## exact p-value with ties##
## Wilcoxon rank sum test with continuity correction
##
## data: x and y
## W = 24, p-value = 0.01392
## alternative hypothesis: true location shift is not equal to 0The small \(p\)-value (0.0139) indicates rejection of the null hypothesis at significance level 0.05. We conclude that the data provide sufficient evidence that the distributions of wing stroke frequencies for the two species differ.
Note: R gives warning messages when there is any tie from the two samples.
Chapter 6. Kruskal-Wallis Test (or H Test)
The Kruskal-Wallis test is a generalization of the Mann-Whitney U test as a nonparametric alternative to the One Way ANOVA based on two or more independent samples.
Review of the Traditional One-Way Analysis of Variance (ANOVA)
Refer to this page: https://datatab.net/tutorial/one-factorial-anova
Kruskal-Wallis Test
The steps of the Kruskal-Wallis test are as follows:
Step 1. Pool values from all samples and find the rank of each value. If two or more values are tied for the same rank, then the average of the ranks that would have been assigned to these values is assigned to each of these tied values.
Step 2. Calculate the treatment sum of squares using the ranks rather than the actual observations. That is, calculate
\[V=\sum_{i=1}^{k}n_i (\bar{R}_i-\bar{R})^2\]
where \(k\) is the number of groups, \(n_i\) is the sample size of the \(i\)th sample, \(\bar{R}_i\) is the mean rank of values in the \(i\)th sample, and \(\bar{R}\) is the mean rank of all observations. Suppose the total sample size is \(n=n_1+n_2+\cdots +n_k\). Since the sum of ranks of all the \(n\) observations equals \(1+2+3+\cdots+n=\frac{n(n+1)}{2}\), \(\bar{R}\) must equal \(\frac{n+1}{2}\). That is,
\[V=\sum_{i=1}^{k}n_i (\bar{R}_i-\frac{n+1}{2})^2\]
Step 3. Kruskal-Wallis (1952) considered the test statistic
\(H=\frac{12V}{n(n+1)}\) and it can be shown that \[H=\frac{12}{n(n+1)}\sum_{i=1}^{k}\frac{R_i^2}{n_i}-3(n+1)\]
Step 4. The \(p\)-value can be calculated using a Chi-square table with \(k-1\) degrees of freedom.
The R code for doing Kruskal-Wallis test is
\[kruskal.test(list(x, y, z, ...))\]
where \(x, y, z, \cdots\) are samples. If the data are in a data frame, the R code would be
\[kruskal.test(y\sim group, data = myData)\] where \(y\) is a quantitative variable and \(group\) is a variable taking values that represent different groups.
A nice video on Kruskal-Wallis test: https://www.youtube.com/watch?v=l86wEhUzkY4
Theory on Kruskal-Wallis test: http://mathcenter.oxford.emory.edu/site/math117/nonParametricTests/
Example 1.
Given 3 samples:
normal subjects- 2.9, 3.0, 2.5, 2.6, 3.2
with obstructive airway disease- 3.8, 2.7, 4.0, 2.4
with asbestosis- 2.8, 3.4, 3.7, 2.2, 2.0
Calculate the value of the Kruskal-Wallis test statistic by hand.
Use the traditional ANOVA method and the Kruskal-Wallis method to test whether the means of the corresponding populations differ.
Solution.
\[n_1=5, n_2=4, n_3=5, n = n_1+n_2+n_3=14\]
Sort ALL observations from smallest to largest: \[2.0, 2.2, 2.4, 2.5, 2.6, 2.7, 2.8, 2.9, 3.0, 3.2, 3.4, 3.7, 3.8, 4.0\] We table the data along with their ranks:
Table for Kruskal-Wallis Test
y
group
rank
2.9
normal
8
3
normal
9
2.5
normal
4
2.6
normal
5
3.2
normal
10
3.8
obstructive airway disease
13
2.7
obstructive airway disease
6
4
obstructive airway disease
14
2.4
obstructive airway disease
3
2.8
asbestosis
7
3.4
asbestosis
11
3.7
asbestosis
12
2.2
asbestosis
2
2
asbestosis
1
The sum of the ranks for sample 1 is: \(R_1=8+9+4+5+10=36\)
The sum of the ranks for sample 2 is: \(R_2=13+6+14+3=36\)
The sum of the ranks for sample 3 is: \(R_3=7+11+12+2+1=33\)
The value of the Kruskal-Wallis test statistic is \[H=\frac{12}{n(n+1)}\sum_{i=1}^{k}\frac{R_i^2}{n_i}-3(n+1)=\frac{12}{14(14+1)}(\frac{36^2}{5}+\frac{36^2}{4}+\frac{33^2}{5})-3(14+1)=0.7714286\]
The R code is
x <- c(2.9, 3.0, 2.5, 2.6, 3.2) # normal subjects
y <- c(3.8, 2.7, 4.0, 2.4) # with obstructive airway disease
z <- c(2.8, 3.4, 3.7, 2.2, 2.0) # with asbestosis
# Doing the Kruskal-Wallis test
kruskal.test(list(x, y, z))##
## Kruskal-Wallis rank sum test
##
## data: list(x, y, z)
## Kruskal-Wallis chi-squared = 0.77143, df = 2, p-value = 0.68# Traditional ANOVA method
D = data.frame(v = c(x, y, z), group = rep(c("normal", "obstructive airway disease", "asbestosis"), c(5, 4, 5)))
D## v group
## 1 2.9 normal
## 2 3.0 normal
## 3 2.5 normal
## 4 2.6 normal
## 5 3.2 normal
## 6 3.8 obstructive airway disease
## 7 2.7 obstructive airway disease
## 8 4.0 obstructive airway disease
## 9 2.4 obstructive airway disease
## 10 2.8 asbestosis
## 11 3.4 asbestosis
## 12 3.7 asbestosis
## 13 2.2 asbestosis
## 14 2.0 asbestosisresult = aov(v ~ group, data = D)
summary(result)## Df Sum Sq Mean Sq F value Pr(>F)
## group 2 0.447 0.2234 0.56 0.587
## Residuals 11 4.387 0.3989The \(p\)-value based on the Kruskal-Wallis method is not small (0.68), so we do not reject the null hypothesis and conclude that the data do not provide sufficient evidence that the samples come from populations with different means.
The \(p\)-value based on the traditional ANOVA method is not small (0.587), so we do not reject the null hypothesis and conclude that the data do not provide sufficient evidence that the samples come from populations with different means.
Example 2.
Use the “iris” data available from R to test whether the variable “sepal length” has different means for different species. Use both the traditional ANOVA method and the Kruskal-Wallis method.
# Test whether Sepal Length of the iris data differ for different species
kruskal.test(Sepal.Length~Species, data = iris)##
## Kruskal-Wallis rank sum test
##
## data: Sepal.Length by Species
## Kruskal-Wallis chi-squared = 96.937, df = 2, p-value < 2.2e-16# Traditional ANOVA method
result = aov(Sepal.Length~Species, data = iris)
summary(result)## Df Sum Sq Mean Sq F value Pr(>F)
## Species 2 63.21 31.606 119.3 <2e-16 ***
## Residuals 147 38.96 0.265
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Both method give an extreme small value (< 2.2e-16), so we reject the null hypothesis and conclude that the data provide sufficient evidence that the variable “sepal length” has different means for different species.
To find which means are different, we can do multiple testing with a p-value adjustment method, such as the Bonferroni correction method:
#perform pairwise t-tests with Bonferroni's correction
pairwise.t.test(x = iris$Sepal.Length, g = iris$Species, p.adjust.method="bonferroni")##
## Pairwise comparisons using t tests with pooled SD
##
## data: iris$Sepal.Length and iris$Species
##
## setosa versicolor
## versicolor 2.6e-15 -
## virginica < 2e-16 8.3e-09
##
## P value adjustment method: bonferroniThe result shows that each group is significantly different.
Post-hoc Tests for Multiple Comparisons Following the Kruskal-Wallis Test
If the Kruskal-Wallis test shows a significant difference among groups that are compared, we can perform the pairwise Wilcoxon test to find which groups are different from each other, possibly with a correction (such as the Bonferroni correction) to control the experiment-wise error rate (i.e. \(\alpha\)).
The Bonferroni correction works like this: If the significance level is chosen to be \(\alpha\), then compare each \(p\)-value with \(\frac{\alpha}{k}\), where \(k\) is the number of comparisons made. Software does it automatically if you set p.adjust.method = “bonferroni”.
pairwise.wilcox.test(iris$Sepal.Length, iris$Species, p.adjust.method = "bonferroni")##
## Pairwise comparisons using Wilcoxon rank sum test with continuity correction
##
## data: iris$Sepal.Length and iris$Species
##
## setosa versicolor
## versicolor 2.5e-13 -
## virginica < 2e-16 1.8e-06
##
## P value adjustment method: bonferroniLet’s take the experiment-wise error rate (i.e. \(\alpha\)) to be 0.05. Since all 3 \(p\)-values are less than \(\frac{\alpha}{k}\) or 0.0167, all species differ in sepal length.
Chapter 7. Friedman Test
The Kruskal-Wallis test handles the situation where there are \(k\) independent samples, one from each of \(k\) populations. The method can be used for comparing \(k\)treatments in the ordinary one-way ANOVA design.
Subjects can vary greatly in many aspects such as in age, health, and many other traits, and thus the responses of subjects who receive the same treatment can also vary greatly. The ordinary one-way ANOVA design fails to control those extraneous factors.
Review of Randomized Complete Block Design (RCBD)
To control the variation in the response due to extraneous factors, a randomized complete block design can be used to compare treatments of interest. In such a design, each of \(b\) blocks receives each of \(k\) treatments only once in random order. For an experiment that involve human subjects, the blocks may be created by age (or IQ) if the effects of treatments differ by age (or IQ). For an agricultural experiment, the blocks may be created by location if the effects of treatments differ by location. For an industrial experiment, the blocks may be created by batch if the effects of treatments differ by batch. Here, the variables “age”, “location”, and “batch” are called blocking factors. These factors should be controlled when comparing treatments. The ordinary one-way ANOVA design fails to control these factors and is thus less powerful.
The RCBD allows us to separate the variation due to the effect of the blocking factor from the effect of the treatment. As a result, the sensitivity of the experiment to detect real differences between treatments is increased (i.e., increase the power of the test). Animal experiments in medicine often use animal litters as blocks, applying all the treatments, one each, to animals within a litter. Because of heredity, animals within a litter are more homogeneous than between litters. This type of blocking reduces litter-to-litter variation.
A special RCBD, called the repeated measures design, is a design in which each subject is measured multiple times to see changes to an intervention (or receives all \(k\) treatments in random order at different time points). Here, each subject is used as a block.
Given a randomized complete block design with \(b\) blocks and \(k\) treatments or a repeated measures design with \(b\) subjects and \(k\) treatments, we make the following assumptions:
The \(b\) blocks are mutually independent, so observations between blocks are independent.
The measurement scale is at least ordinal within each block.
There is no interaction between blocks and treatments.
Traditional Analysis of Data Based on a Randomized Complete Block Design
Example 1.

Is there any evidence of a difference in mean response among the three treatments?
Solution.
Hand calculation: https://stattrek.com/anova/randomized-block/example
Find treatment means:
Mean of treatment 1 = \((67+67.5+76+72.7+73.1+65.8+75.6)/7=71.1\). Mean of treatment 2 = \((71.9+68.8+82.6+78.1+74.2+70.8+84.9)/7=75.9\). Mean of treatment 3 = \((72.2+66.4+74.5+67.3+73.2+68.7+69)/7=70.1857\).
Find block means:
Mean of Block 1 = \((67+71.9+72.2)/3=70.3667\). Mean of Block 2 = \((67.5+68.8+66.4)/3=67.5667\). Mean of Block 3 = \((76+82.6+74.5)/3=77.7\). Mean of Block 4 = \((72.7+78.1+67.3)/3=72.7\). Mean of Block 5 = \((73.1+74.2+73.2)/3=73.5\). Mean of Block 6 = \((65.8+70.8+68.7)/3=68.4333\). Mean of Block 7 = \((75.6+84.9+69)/3=76.5\).
The grand mean: \((71.1+75.9+70.1857)/3=72.3952\).
The total sum of squares: \(SST = (67-72.3952)^2+(67.5-72.3952)^2+\cdots+(68.7-72.3952)^2+(69-72.3952)^2=524.6495\)
The sum of squares for treatments: \(SS_{treatment} = 7\cdot[(70.3667-72.3952)^2 + (75.9-72.3952)^2 + (70.1857-72.3952)^2] = 131.9014\)
The sum of squares for blocks: \[SS_{block} = 3\cdot[(70.36667-72.3952)^2 + (67.5667-72.3952)^2 + (77.7-72.3952)^2 + (72.7-72.3952)^2\] \[+ (73.5-72.3952)^2+ (68.4333-72.3952)^2+ (76.5-72.3952)^2] = 268.2894\]
The error sum of squares: \((67-71.1-70.3667+72.3952)^2 + (71.9-75.9-70.3667+72.3952)^2 + \cdots + (69-70.1857-76.5+72.3952)^2 =124.4481\)
We can see that \[SST=SS_{treatment}+SS_{block}+SS_{error}\] The R code for analyzing data based on a RCBD:
Block = gl(7, 3, 7*3) # Produce 1 1 1 2 2 2 3 3 3 4 4 4 5 5 5 6 6 6 7 7 7 with 7 levels
Treatment = rep(c("A", "B", "C"), 7)
y = c(67, 71.9, 72.2, 67.5, 68.8, 66.4, 76, 82.6, 74.5, 72.7, 78.1, 67.3, 73.1, 74.2, 73.2, 65.8, 70.8, 68.7, 75.6, 84.9, 69)
D = data.frame(Block, Treatment, y)
fit <- aov(y ~ Block + Treatment, data = D)
summary(fit)## Df Sum Sq Mean Sq F value Pr(>F)
## Block 6 268.3 44.71 4.311 0.0151 *
## Treatment 2 131.9 65.95 6.359 0.0131 *
## Residuals 12 124.5 10.37
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1# We can also use a regression approach
myModel <- lm(y ~ Block + Treatment, data = D)
anova(myModel) # Same result## Analysis of Variance Table
##
## Response: y
## Df Sum Sq Mean Sq F value Pr(>F)
## Block 6 268.29 44.715 4.3113 0.01509 *
## Treatment 2 131.90 65.950 6.3588 0.01309 *
## Residuals 12 124.46 10.372
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Here is an explanation of the result:
The effects of different treatments are significantly different (p-value = 0.0151) at the significance level 0.05.
We can create a set of confidence intervals on the differences between the means of the levels of the treatment (or the factor of interest) with the specified family-wise error rate (i.e., confidence level or probability of coverage). Here is a tutorial regarding the family-wise error rate: https://www.statology.org/family-wise-error-rate/.
To get 95% family-wise confidence intervals for the mean differences based on Tukey’s multiple comparisons of means, do
# Compute Tukey's Honest Significant Differences
TukeyHSD(aov(myModel), which = "Treatment", conf.level = 0.95)## Tukey multiple comparisons of means
## 95% family-wise confidence level
##
## Fit: aov(formula = myModel)
##
## $Treatment
## diff lwr upr p adj
## B-A 4.8000000 0.2074677 9.392532 0.0404168
## C-A -0.9142857 -5.5068180 3.678247 0.8577520
## C-B -5.7142857 -10.3068180 -1.121753 0.0156234The result also reports p-values for tests between treatment means. Of course, these \(p\)-values do not depend on the set-up of the confidence level.
The intervals can be plotted:
# Compute Tukey's Honest Significant Differences
tukeyIntervals = TukeyHSD(aov(myModel), which = "Treatment", conf.level = 0.95)
plot(tukeyIntervals)
Introduction to One-Way Repeated Measures ANOVA
When there are \(n\) individuals and each individual is measured repeatedly at \(k\) time points or is observed under \(k\) conditions, we can use the one-way repeated measures ANOVA. The purpose is to determine whether there is any difference in the response variable at different time points or under different conditions. A short introduction: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4320283/
Example 1.
Five participants of a program for weight loss reported their weight once a week for a total of 3 weeks.
D = data.frame(participant = rep(1:5, c(3,3,3,3,3)),
week = rep(1:3, 5),
weight = c(160, 155, 152, 178, 177, 174, 182, 183, 175, 162, 158, 160, 175, 173, 162))
D$participant = factor(D$participant)
D$week = factor(D$week)
kbl(D, table.attr = "style='width:60%; '") %>%
kable_classic()| participant | week | weight |
|---|---|---|
| 1 | 1 | 160 |
| 1 | 2 | 155 |
| 1 | 3 | 152 |
| 2 | 1 | 178 |
| 2 | 2 | 177 |
| 2 | 3 | 174 |
| 3 | 1 | 182 |
| 3 | 2 | 183 |
| 3 | 3 | 175 |
| 4 | 1 | 162 |
| 4 | 2 | 158 |
| 4 | 3 | 160 |
| 5 | 1 | 175 |
| 5 | 2 | 173 |
| 5 | 3 | 162 |
Is there sufficient evidence to conclude that there are differences in mean weight in different weeks? Use \(\alpha=0.05\).
Solution.
We use the parametric method introduced here: https://www.datanovia.com/en/lessons/repeated-measures-anova-in-r/
The null hypothesis \(H_0: \mu_1 = \mu_2 = \mu_3\) (the mean weights are all equal)
The alternative hypothesis: \(H_a: \text{at least one mean is different from the rest}\)
Compute summary statistics:
library(datarium)
library(rstatix)
D %>%
group_by(week) %>%
get_summary_stats(weight, type = "mean_sd")## # A tibble: 3 x 5
## week variable n mean sd
## <fct> <chr> <dbl> <dbl> <dbl>
## 1 1 weight 5 171. 9.84
## 2 2 weight 5 169. 12.2
## 3 3 weight 5 165. 9.79Boxplots for different weeks:
library(ggpubr)
ggboxplot(D, x = "week", y = "weight", add = "point")
Check normality by Shapilo-Wilk test:
D %>%
group_by(week) %>%
shapiro_test(weight)## # A tibble: 3 x 4
## week variable statistic p
## <fct> <chr> <dbl> <dbl>
## 1 1 weight 0.882 0.320
## 2 2 weight 0.908 0.453
## 3 3 weight 0.909 0.463Create QQ plots for checking normality:
ggqqplot(D, "weight", facet.by = "week")
Fit a One-Way Repeated Measures ANOVA model:
library(datarium)
library(rstatix)
res.aov<-anova_test(data = D,
dv = weight, # Dependent/response variable
wid = participant, # Block
within = week # 3 observations are for each participant
)
get_anova_table(res.aov)## ANOVA Table (type III tests)
##
## Effect DFn DFd F p p<.05 ges
## 1 week 2 8 7.417 0.015 * 0.081The weight is significantly different in different weeks, with \(F(2, 8) = 7.417, ~p = 0.0.0151\), and effect size (generalized eta squared) \(ges=0.081\), where,
\(F\) Indicates that we are comparing to an \(F\)-distribution (\(F\)-test); (2, 8) indicates the degrees of freedom in the numerator (DFn) and the denominator (DFd), respectively;
\(p\) specifies the \(p\)-value.
\(ges\) is the generalized effect size (amount of variability due to the within-subjects factor).
We can also use the base function “aov()” to analyze data from a repeated measures design.
# fit repeated measures ANOVA model
model <- aov(weight ~ week + Error(participant), data = D)
summary(model)##
## Error: participant
## Df Sum Sq Mean Sq F value Pr(>F)
## Residuals 4 1298 324.6
##
## Error: Within
## Df Sum Sq Mean Sq F value Pr(>F)
## week 2 120.40 60.20 7.417 0.0151 *
## Residuals 8 64.93 8.12
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1# or simply
model <- aov(weight ~ week + participant, data = D)
summary(model)## Df Sum Sq Mean Sq F value Pr(>F)
## week 2 120.4 60.2 7.417 0.0151 *
## participant 4 1298.3 324.6 39.988 2.48e-05 ***
## Residuals 8 64.9 8.1
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1# The second method produce more information, allowing us to calculate the generalized eta square
120.4/(120.4+1298.3+64.9) # gives same value 0.081 as before## [1] 0.08115395The result is the same as that obtained by using “anova_test” from package “rstatix”.
Report of results:
A one-way repeated measures ANOVA was conducted on five children to examine the effect that four antibiotics had on ratings of the tastes of the antibiotics. The results showed that the type of antibiotic used lead to no statistically significant differences in the rating of taste (F(2, 8) = 7.417, p = 0.0151).
Example 2.
In a study of palatability of antibiotics for children, the researchers used a voluntary sample of 5 healthy children to assess their reactions to the taste of four antibiotics. The children’s responses were measured on a 10-centimeter visual analog scale that incorporated the use of faces, from sad (low score) to happy (high score). The minimum and maximum scores were, respectively, 0 and 10. The data below were obtained when each of 5 children were asked to rate the taste of all 4 antibiotics.
|
Antibiotic
|
||||
|---|---|---|---|---|
| Children | I | II | III | IV |
| 1 | 4.8 | 2.2 | 6.8 | 6.2 |
| 2 | 8.1 | 9.2 | 6.6 | 9.6 |
| 3 | 5.0 | 2.6 | 3.6 | 6.5 |
| 4 | 7.9 | 9.4 | 5.3 | 8.5 |
| 5 | 3.9 | 7.4 | 2.1 | 2.0 |
| Note: | ||||
| Data from Mathematical Statistics with applications by Wackerly et. al. | ||||
Is there sufficient evidence to conclude that there are differences in the perceived taste of the different antibiotics? Use \(\alpha=0.05\).
Why did the researcher have each child rank all four antibiotics instead of using the ordinary one-way ANOVA design (that is, using 20 different children, randomly selecting 5 to receive only antibiotic I, another 5 to receive only antibiotic II, and so on)?
Solution.
We use the parametric method introduced here: https://www.datanovia.com/en/lessons/repeated-measures-anova-in-r/
The null hypothesis \(H_0: \mu_1 = \mu_2 = \mu_3 = \mu_4\) (the treatment means are all equal)
The alternative hypothesis: \(H_a: \text{at least one treatment mean is different from the rest}\)
To analyze the data, we must first convert it into long format:
library(datarium)
library(rstatix)
Children = gl(5, 4, 5*4)
antibiotic = rep(c("I", "II", "III", "IV"), 5)
rating = c(4.8, 2.2, 6.8, 6.2, 8.1, 9.2, 6.6, 9.6, 5.0, 2.6, 3.6, 6.5, 7.9, 9.4, 5.3, 8.5, 3.9, 7.4, 2.1, 2.0)
D = data.frame(Children, antibiotic, rating)
D## Children antibiotic rating
## 1 1 I 4.8
## 2 1 II 2.2
## 3 1 III 6.8
## 4 1 IV 6.2
## 5 2 I 8.1
## 6 2 II 9.2
## 7 2 III 6.6
## 8 2 IV 9.6
## 9 3 I 5.0
## 10 3 II 2.6
## 11 3 III 3.6
## 12 3 IV 6.5
## 13 4 I 7.9
## 14 4 II 9.4
## 15 4 III 5.3
## 16 4 IV 8.5
## 17 5 I 3.9
## 18 5 II 7.4
## 19 5 III 2.1
## 20 5 IV 2.0Compute summary statistics:
D %>%
group_by(antibiotic) %>%
get_summary_stats(rating, type = "mean_sd")## # A tibble: 4 x 5
## antibiotic variable n mean sd
## <chr> <chr> <dbl> <dbl> <dbl>
## 1 I rating 5 5.94 1.93
## 2 II rating 5 6.16 3.52
## 3 III rating 5 4.88 2.01
## 4 IV rating 5 6.56 2.91Boxplots for different antibiotics:
library(ggpubr)
ggboxplot(D, x = "antibiotic", y = "rating", add = "point")
Check normality by Shapilo-Wilk test:
D %>%
group_by(antibiotic) %>%
shapiro_test(rating)## # A tibble: 4 x 4
## antibiotic variable statistic p
## <chr> <chr> <dbl> <dbl>
## 1 I rating 0.844 0.175
## 2 II rating 0.820 0.116
## 3 III rating 0.915 0.496
## 4 IV rating 0.926 0.571Create QQ plots for checking normality:
ggqqplot(D, "rating", facet.by = "antibiotic")
Fit a One-Way Repeated Measures ANOVA model:
res.aov<-anova_test(data = D,
dv = rating, # Dependent/response variable
wid = Children, # Block
within = antibiotic # Treatments are given within each child
)
get_anova_table(res.aov)## ANOVA Table (type III tests)
##
## Effect DFn DFd F p p<.05 ges
## 1 antibiotic 3 12 0.653 0.596 0.063The rating is significantly different in the perceived taste of the different antibiotics, with \(F(3, 12) = 0.653, ~p = 0.596\), and effect size (generalized eta squared) \(ges=0.063\), where,
\(F\) Indicates that we are comparing to an \(F\)-distribution (\(F\)-test); (3, 12) indicates the degrees of freedom in the numerator (DFn) and the denominator (DFd), respectively;
\(p\) specifies the \(p\)-value.
\(ges\) is the generalized effect size (amount of variability due to the within-subjects factor).
We can also use the base function “aov()” to analyze data from a repeated measures design.
# fit repeated measures ANOVA model
model <- aov(rating ~ antibiotic + Error(Children), data = D)
summary(model)##
## Error: Children
## Df Sum Sq Mean Sq F value Pr(>F)
## Residuals 4 67.31 16.83
##
## Error: Within
## Df Sum Sq Mean Sq F value Pr(>F)
## antibiotic 3 7.72 2.574 0.653 0.596
## Residuals 12 47.29 3.941# or simply
model <- aov(rating ~ antibiotic + Children, data = D)
summary(model)## Df Sum Sq Mean Sq F value Pr(>F)
## antibiotic 3 7.72 2.574 0.653 0.5962
## Children 4 67.31 16.828 4.270 0.0223 *
## Residuals 12 47.29 3.941
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1# The second method produce more information, allowing us to calculate the generalized eta square
7.72/(7.72+67.31+47.29) # gives same value 0.063 as before## [1] 0.06311315The result is the same as that obtained by using “anova_test” from package “rstatix”.
Report of results:
A one-way repeated measures ANOVA was conducted on five children to examine the effect that four antibiotics had on ratings of the tastes of the antibiotics. The results showed that the type of antibiotic used lead to no statistically significant differences in the rating of taste (F(3, 12) = 0.653, p = 0.596).
Post-Hoc Tests for Repeated Measures ANOVA
If a one-way repeated measures ANOVA shows any significance difference in the mean response, post-hoc tests can be performed using the pairwise t-test method. Different methods for correcting the p-values of multiple tests can be used.
The weight loss example in the previous section indicated that weight differs significantly in different weeks. We perform post-hoc tests below.
D = data.frame(participant = rep(1:5, c(3,3,3,3,3)),
week = rep(1:3, 5),
weight = c(160, 155, 152, 178, 177, 174, 182, 183, 175, 162, 158, 160, 175, 173, 162))
D$participant = factor(D$participant)
D$week = factor(D$week)
pairwise.t.test(D$weight,
g = D$week, # g means group
paired = TRUE, # This is key!
p.adjust.method = "bonferroni"
)##
## Pairwise comparisons using paired t tests
##
## data: D$weight and D$week
##
## 1 2
## 2 0.325 -
## 3 0.067 0.331
##
## P value adjustment method: bonferroni# Or
pairwise_t_test(weight ~ week, data = D,
paired = TRUE,
p.adjust.method = "bonferroni")## # A tibble: 3 x 10
## .y. group1 group2 n1 n2 statistic df p p.adj p.adj.signif
## * <chr> <chr> <chr> <int> <int> <dbl> <dbl> <dbl> <dbl> <chr>
## 1 weight 1 2 5 5 2.06 4 0.108 0.324 ns
## 2 weight 1 3 5 5 3.61 4 0.022 0.068 ns
## 3 weight 2 3 5 5 2.04 4 0.11 0.33 nsAs the result shows, at significance level 0.05, no pair-wise comparison is significant. You might feel this is strange since the overall test is significant but no pairwise comparison is significant. The reason is that the Bonferroni method is conservative and tends to give nonsignificant results.
Advantages with Running a Repeated-Measures ANOVA over an Ordinary ANOVA
The major advantage with running a repeated measures ANOVA over an ordinary ANOVA is that the test is generally much more powerful. This particular advantage is achieved by the reduction in the mean square error that comes from the partitioning of variability due to differences between blocks (subjects) from the original error term in an ordinary ANOVA. This is seen from doing an ordinary ANOVA for the data:
m=aov(weight~week, data=D)
summary(m)## Df Sum Sq Mean Sq F value Pr(>F)
## week 2 120.4 60.2 0.53 0.602
## Residuals 12 1363.2 113.6The \(p\)-value is larger when doing an ordinary ANOVA.
The Friedman Test: The Nonparametric Alternative to One-Way Repeated Measures ANOVA
The Friedman test is the nonparametric alternative to the one-way repeated measures ANOVA. It is a rank sum method.
Suppose there are \(b\) blocks and \(k\) treatments. The steps of doing a Friedman test are:
Step 1. State the null hypothesis and alternative hypothesis. \[H_0: \text{The effects of all treatments are the same.}\] \[H_a: \text{The effects of all treatments are not all same.}\]
Step 2. Calculate the value of the test statistic \(F_r\) (also called \(Q\)). This is done in two sub-steps:
Step 2.1. Rank the values in each block separately, with tied values receiving the average rank of ranks that would be assigned to them.
Step 2.2. Compute the sum of ranks for each treatment group.
\[F_r = \frac{12}{bk(k+1)}\sum_{i=1}^{k}R_i^2-3b(k+1)\] or \[F_r = \frac{(k-1)\sum_{i=1}^{k}[R_i-\frac{b(k+1)}{2}]^2}{A-C}\]
when there is any tie, where \(R_i\) is the sum of ranks for group \(i\), \(i=1,2,\cdots k\), A is the sum of all squared ranks and \(C=\frac{bk(k+1)^2}{4}\).
Step 3. Calculate the \(p\)-value.
Step 4. Make a decision and draw a conclusion.
A nice video on Friedman Test: https://www.youtube.com/watch?v=2moNzzkkZwU and for more detail on hand calculation: https://datatab.net/tutorial/friedman-test
Example 3.
This is a continuation to the Example 2. Use the Friedman test to answer the question.
Solution.
Children = gl(5, 4, 5*4)
antibiotic = rep(c("I", "II", "III", "IV"), 5)
rating = c(4.8, 2.2, 6.8, 6.2, 8.1, 9.2, 6.6, 9.6, 5.0, 2.6, 3.6, 6.5, 7.9, 9.4, 5.3, 8.5, 3.9, 7.4, 2.1, 2.0)
D = data.frame(Children, antibiotic, rating)
res.fried = friedman.test(rating ~ antibiotic|Children, data = D)
res.fried##
## Friedman rank sum test
##
## data: rating and antibiotic and Children
## Friedman chi-squared = 1.56, df = 3, p-value = 0.6685From the output of the Friedman test, we know that there is no significant difference between groups.
Post-hoc Tests Following a Significant Friedman Test Result
We can perform post-hoc tests following a significant Friedman test result, possibly with a correction (such as the Bonferroni correction) to control the experiment-wise error rate. These tests are pairwise Wilcoxon signed-rank tests for identifying which groups are different. P-values can be adjusted for multiple comparisons.
We will do the follow-up test using the antibiotics example only for showing how it can be done.
# pairwise comparisons
pwc <- D %>%
wilcox_test(rating ~ antibiotic, paired = TRUE, p.adjust.method = "bonferroni")
pwc## # A tibble: 6 x 9
## .y. group1 group2 n1 n2 statistic p p.adj p.adj.signif
## * <chr> <chr> <chr> <int> <int> <dbl> <dbl> <dbl> <chr>
## 1 rating I II 5 5 7 1 1 ns
## 2 rating I III 5 5 11 0.438 1 ns
## 3 rating I IV 5 5 5 0.588 1 ns
## 4 rating II III 5 5 10 0.625 1 ns
## 5 rating II IV 5 5 7 1 1 ns
## 6 rating III IV 5 5 3 0.312 1 nsAs expected, no pairwise difference is statistically significant and thus no need to do post-hoc tests.
Example 4.
Each of 6 technicians is performing 3 tasks (A, B, and C) in random order. The completion times are given below:
| Technician | Task A | Rank | Task B | Rank | Task C | Rank |
| 1 | 1.21 | 1.56 | 1.48 | |||
| 2 | 1.63 | 2.01 | 1.63 | |||
| 3 | 1.42 | 1.7 | 2.06 | |||
| 4 | 1.16 | 1.27 | 1.27 | |||
| 5 | 2.43 | 2.64 | 1.98 | |||
| 6 | 1.94 | 2.81 | 2.44 | |||
| Note: | ||||||
| Reference: Data from Mathematical Statistics with Applications by Wackerly et. al |
The R code for Friedman test:
D = data.frame(Technician = rep(1:6, 3),
Task = rep(c("A", "B", "C"), c(6, 6, 6)),
Time = c(1.21, 1.63, 1.42, 1.16, 2.43, 1.94,
1.56, 2.01, 1.70, 1.27, 2.64, 2.81,
1.48, 1.63, 2.06, 1.27, 1.98, 2.44))
res.fried = friedman.test(Time ~ Task|Technician, data = D)
res.fried##
## Friedman rank sum test
##
## data: Time and Task and Technician
## Friedman chi-squared = 7.3636, df = 2, p-value = 0.02518From the output of the Friedman test, we know that there is a significant difference between groups (Chi-squared = 7.3636, p-value = 0.0252).
Note: If you find the value of the Friedman test statistic, you will get 6.75, which is not equal to 7.3636. This is due to some corrections by the algorithm adopted by R.
Since there is a significant difference between groups, we do the post-hoc test below:
# pairwise comparisons
pwc <- D %>%
wilcox_test(Time ~ Task, paired = TRUE, p.adjust.method = "bonferroni")
pwc## # A tibble: 3 x 9
## .y. group1 group2 n1 n2 statistic p p.adj p.adj.signif
## * <chr> <chr> <chr> <int> <int> <dbl> <dbl> <dbl> <chr>
## 1 Time A B 6 6 0 0.031 0.094 ns
## 2 Time A C 6 6 3 0.281 0.843 ns
## 3 Time B C 6 6 13 0.178 0.534 nsThe result shows no treatment is significantly different from any other one after Bonferroni correction.
Chapter 8. Rank Correlation Tests
The ordinary Correlation between Two Quantitative Variables
Suppose there are \(n\) pairs of data \((x_i, y_i)\), \(i=1,2,\cdots, n\). The ordinary Pearson correlation coefficient between two quantitative variables \(X\) and \(Y\) is defined by
\[r = \frac{\sum_{i=1}^{n}(x_i-\bar{x})(y_i-\bar{y})}{\sqrt{\sum_{i=1}^{n}(x_i-\bar{x})^2}\cdot\sqrt{\sum_{i=1}^{n}(y_i-\bar{y})^2}}=\frac{\sum_{i=1}^{n}x_iy_i-\frac{1}{n}(\sum_{i=1}^{n}x_i)(\sum_{i=1}^{n}y_i)}{\sqrt{\sum_{i=1}^{n}x_i^2-\frac{1}{n}(\sum_{i=1}^{n}x_i)^2}\cdot\sqrt{ \sum_{i=1}^{n}y_i^2-\frac{1}{n}(\sum_{i=1}^{n}y_i)^2 }}\]
To test the Pearson correlation between two quantitative variables, the null and alternative hypotheses are \(H_0:\rho=0\) vs. \(H_0:\rho<0\) or \(H_0:\rho>0\) or \(H_0:\rho\ne0\).
The test statistic is
\[t=\frac{r}{\sqrt{1-r^2}}\cdot\sqrt{n-2}\] under the null hypothesis, the test statistic has a t-distribution with \(n-2\) degrees of freedom.
The R code for testing Pearson correlation is
- cor.test(x, y, method = “pearson”)
Spearman Rank Correlation Test
Spearman correlation is often used to evaluate the relationship between two ordinal variables. Examples of ordinal variables include
Course grade (A, B, C, D, F),
Level of satisfaction (Extremely satisfied, Very satisfied, Somewhat satisfied, Somewhat dissatisfied, Very dissatisfied, and Extremely dissatisfied), and
Level of agreement (Strongly Disagree, Disagree, Neutral, Agree, Strongly Agree).
The Spearman correlation is calculated using the same formula as used for Pearson correlation, but by using ranks of values for each variable. The ranks are determined separately for each of the two variables involved.
When there is no tie, a simplified formula is available:
\[r_s=1-\frac{6\Sigma d^2}{n(n^2-1)}\]
The R code for calculating and testing hypotheses for the above correlations is:
\[cor(x,~y, ~method=c("pearson", "kendall", "spearman"))\] \[cor.test(x, ~y, ~method=c("pearson", "kendall", "spearman"))\] ### Examples
Example 1.
x = c(3, 7, 10, 15, 20, 30, 35)
y = c(8, 18, 20, 12, 38, 34, 50)
plot(y~x)
# Calculate Pearson correlation by hand
n = length(x)
(sum(x*y)-sum(x)*sum(y)/n)/(sqrt(sum(x^2)-sum(x)^2/n)*sqrt(sum(y^2)-sum(y)^2/n))## [1] 0.8915852# Calculate Pearson correlation
cor(x, y, method = "pearson") ## [1] 0.8915852# Calculate Spearman correlation
cor(x, y, method = "spearman") ## [1] 0.8571429# Test hypotheses about Pearson correlation
cor.test(x, y, method = "pearson") ##
## Pearson's product-moment correlation
##
## data: x and y
## t = 4.4024, df = 5, p-value = 0.007006
## alternative hypothesis: true correlation is not equal to 0
## 95 percent confidence interval:
## 0.4215874 0.9839823
## sample estimates:
## cor
## 0.8915852# Test hypotheses about Pearson correlation
cor.test(x, y, method = "spearman") ##
## Spearman's rank correlation rho
##
## data: x and y
## S = 8, p-value = 0.02381
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## 0.8571429Kendall Rank Correlation Test
The Kendall rank correlation, commonly referred to as Kendall’s \(\tau\) is a statistic used to measure the ordinal association between two ordinal variables.
For the calculation of Kendall’s \(\tau\), use the reference: https://www.statisticshowto.com/kendalls-tau/
We want to test \(H_0: \text{the two variables are independent}\) vs. \(H_a: \text{the two variables are dependent}\). When there is any tie in at least one of the two variables, the test statistic is different. We consider the case of no tie. Under the null hypothesis, the sampling distribution of \(\tau\) has an expected value of zero. The precise distribution cannot be characterized in terms of common distributions, but may be calculated exactly for small samples; for larger samples, \(\tau\) has an approximately normal distribution with mean 0 and variance \(\frac{2(2n+5)}{9n(n-1)}\), so the test statistic is \(T=\frac{C-D}{\sqrt{\frac{n(n-1)(2n+5)}{18}}}\). The p-value is calculated based on the standard normal distribution.
The R code is
- cor.test(x, y, method = “kendall”)
x = c(3, 7, 10, 15, 20, 30, 35)
y = c(8, 18, 20, 12, 38, 34, 50)
plot(y~x)
# To calculate Kendall's tau
cor(x, y, method = "kendall") ## [1] 0.7142857# Test hypotheses about Pearson correlation
cor.test(x, y, method = "kendall") ##
## Kendall's rank correlation tau
##
## data: x and y
## T = 18, p-value = 0.03016
## alternative hypothesis: true tau is not equal to 0
## sample estimates:
## tau
## 0.7142857Chapter 9. K-S & A-D Tests for Goodness of Fit
We can test whether a sample is from a population that has a particular distribution. Such a test is called a goodness-of-fit test.
We use \(F_0(x)\) to denote the cumulative distribution function (CDF) of the population and \(F_n (x)\) to denote the CDF of the sample.
There are a few approaches for testing goodness of fit. We only introduce the Kolmogorov-Smirnov test and the Anderson-Darling test. Both tests involve a distance between the two functions \(F_n(x)\) and \(F_0(x)\).
Kolmogorov-Smirnov Test for Goodness of Fit Based on One Sample
The distance between the two functions is defined by \[ D = max_x |F_n(x)-F_0(x)|\] The distance is defined as the largest vertical distance between two points one on each curve.

To test whether a sample is from a population that has a particular distribution, the steps are:
Step 1. State the null hypothesis and alternative hypothesis are
\[H_0: F(x)=F_0(x) ~~\text{vs.} ~H_a: F(x)\ne F_0(x)\] where \(F(x)\) is the unknown population distribution.
Step 2. Calculate the value of the test statistic \(D\) under the null hypothesis. Denote this value by \(d\).
Step 3. Determine the \(p\)-value.
Under the null hypothesis, the sampling distribution of \(D\) is complicated, a table such as https://www.statisticshowto.com/wp-content/uploads/2016/07/k-s-test-table-p-value.png is available for estimating the \(p\)-value. Since the larger the \(d\), the more evidence to reject the null hypothesis, so the \(p\)-value of the Kolmogorov-Smirnov test equals the area of the right region beyond \(d\) on the number line and under the curve of the sampling distribution of \(D\).
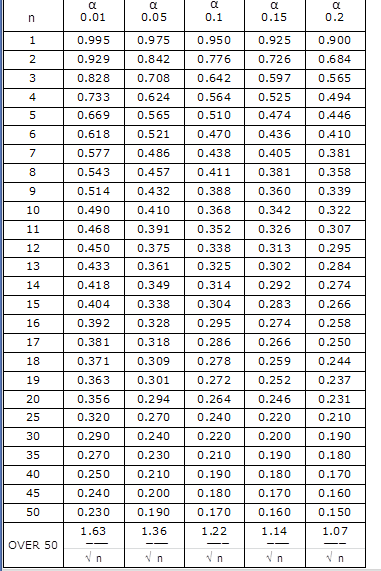{kind=link}
Step 4. Make a decision by comparing this \(p\)-value and a chosen significance level and draw a conclusion.
Example 1.
We observe the following data points:
108.59, 94.42, 96.07, 101.16, 121.20, 97.80, 118.20, 118.51, 96.19, 128.21, 93.22, 85.34, 118.37, 84.16, 101.92, 98.64, 104.13, 115.02, 105.06, 112.79
Is there any evidence to suggest that the data were not randomly sampled from a normal distribution with mean 100 and standard deviation 15?
Solution.
Step 1. The null hypothesis \(H_0\): The distribution is \(N(\mu=100, \sigma=15)\) and the alternative hypothesis \(H_a\): The distribution is not \(N(\mu=100, \sigma=15)\).
Step 2. Construct the following table:
| k | \(y_k\) | \(F_n (y_{k-1})\) | \(F_0 (y_k)\) | \(F_n (y_k)\) | \(F_n (y_{k-1}) - F_0 (y_k)\) | \(F_0 (y_k) - F_n (y_k)\) |
| 1 | 84.16 | 0 | 0.1455 | 0.05 | 0.1455 | 0.0955 |
| 2 | 85.34 | 0.05 | 0.1642 | 0.1 | 0.2256 | 0.0642 |
| 3 | 93.22 | 0.1 | 0.3256 | 0.15 | 0.2256 | 0.1756 |
| 4 | 94.42 | 0.15 | 0.3549 | 0.2 | 0.2049 | 0.1549 |
| 5 | 96.07 | 0.2 | 0.3967 | 0.25 | 0.1967 | 0.1467 |
| 6 | 96.19 | 0.25 | 0.3997 | 0.3 | 0.1497 | 0.0997 |
| 7 | 97.8 | 0.3 | 0.4417 | 0.35 | 0.1417 | 0.0917 |
| 8 | 98.64 | 0.35 | 0.4639 | 0.4 | 0.1139 | 0.0639 |
| 9 | 101.16 | 0.4 | 0.5308 | 0.45 | 0.1308 | 0.0808 |
| 10 | 101.92 | 0.45 | 0.5509 | 0.5 | 0.1009 | 0.0509 |
| 11 | 104.13 | 0.5 | 0.6085 | 0.55 | 0.1085 | 0.0585 |
| 12 | 105.06 | 0.55 | 0.6321 | 0.6 | 0.0821 | 0.0321 |
| 13 | 108.59 | 0.6 | 0.7166 | 0.65 | 0.1166 | 0.0666 |
| 14 | 112.79 | 0.65 | 0.8031 | 0.7 | 0.1531 | 0.1031 |
| 15 | 115.02 | 0.7 | 0.8417 | 0.75 | 0.1417 | 0.0917 |
| 16 | 118.2 | 0.75 | 0.8875 | 0.8 | 0.1375 | 0.0875 |
| 17 | 118.37 | 0.8 | 0.8896 | 0.85 | 0.0896 | 0.0396 |
| 18 | 118.51 | 0.85 | 0.8914 | 0.9 | 0.0414 | 0.0086 |
| 19 | 121.2 | 0.9 | 0.9212 | 0.95 | 0.0212 | 0.0288 |
| 20 | 128.21 | 0.95 | 0.97 | 1 | 0.02 | 0.03 |
| Note: | ||||||
| Reference: <https://online.stat.psu.edu/stat415/lesson/22/22.2>; |
The value of the Kolmogorov-Smirnov test statistic is the largest value among those in the last two columns of the above table, which is 0.2256.
Step 3. To find the \(p\)-value, we use the table https://www.statisticshowto.com/wp-content/uploads/2016/07/k-s-test-table-p-value.png. Since \(n=20\) and \(d = 0.2256\), the \(p\)-value is greater than 0.20 according to the table.
Step 4. Since the \(p\)-value (> 0.20) is greater than the significance level (\(\alpha = 0.05\)), we do not reject the null hypothesis and conclude that the data do not provide sufficient evidence that the population distribution is NOT normal distribution with mean 100 and standard deviation 15.
We can test the hypotheses directly using the following R code:
y = c(108.59, 94.42, 96.07, 101.16, 121.20, 97.80, 118.20, 118.51, 96.19, 128.21, 93.22, 85.34, 118.37, 84.16, 101.92, 98.64, 104.13, 115.02, 105.06, 112.79)
ks.test(y, "pnorm", 100, 15) # Specify the name of the distribution and##
## One-sample Kolmogorov-Smirnov test
##
## data: y
## D = 0.22563, p-value = 0.2239
## alternative hypothesis: two-sided # additional parameters; That is, the null distribution
# need to be fully specified when using the K-S testThe \(p\)-value is 0.2239, which is consistent with the table method.
Kolmogorov-Smirnov Test for Equality of Distributions Based on Two Independent Samples
We can also use the Kolmogorov-Smirnov method to test whether two samples come from the same distribution. This is demonstrated by the following example.
Example.
Test whether the following two samples come from the same distribution.
Sample 1: 83.15, 94.62, 107.82, 95.33, 101.56, 95.45, 81.55, 108.46, 87.92, 100.00, 112.96, 77.90, 99.09, 90.09, 90.49
Sample 2: 107.98, 101.74, 109.20, 132.35, 102.24, 113.55, 118.57, 108.05, 112.56, 128.93, 103.51, 103.05, 81.60, 121.73, 74.10
Solution.
Step 1. The null hypothesis is \(H_0:\) the samples are from the same distribution. The alternative hypothesis is \(H_a:\) the samples are from different distributions.
Step 2. The \(p\)-value can be calculated with the code:
x = c(83.15, 94.62, 107.82, 95.33, 101.56, 95.45, 81.55, 108.46, 87.92, 100.00, 112.96, 77.90, 99.09, 90.09, 90.49)
y = c(107.98, 101.74, 109.20, 132.35, 102.24, 113.55, 118.57, 108.05, 112.56, 128.93, 103.51, 103.05, 81.60, 121.73, 74.10)
plot(ecdf(x), xlim = range(c(x, y)), col = "black",
ylab = expression(F[n](x)),
verticals=TRUE, do.points=FALSE,
main = "Empirical Cumulative Distribution Functions")
plot(ecdf(y), add = TRUE, lty = "dashed", col = "red",
verticals=TRUE, do.points=FALSE)
legend(min(x, y), 0.9, legend = c("Sample 1", "Sample 2"),
text.col = c("black", "red"), bty = "n",
lty = c("solid", "dashed"), col = c("black", "red"))
# Or
df <- data.frame(z = c(x,y),
g = rep(c("Sample 1", "Sample 2"), c(length(x), length(y))))
ggplot(df, aes(z, colour = g)) + stat_ecdf() + labs(color = "Sample")
# KS test of equality of two distributions
ks.test(x, y)##
## Two-sample Kolmogorov-Smirnov test
##
## data: x and y
## D = 0.66667, p-value = 0.001837
## alternative hypothesis: two-sidedThe small p-value (0.0018) indicates there is sufficient evidence that the samples do not come from the same distribution.
Anderson-Darling Test
An alternative to the Kolmogorov-Smirnov test is the Anderson-Darling test. There are a few R packages that perform this test. We only introduce the R package “goftest” (goodness of fit).
Example.
Test whether the following sample
120.8, 99, 104, 87.8, 121.7, 85.9, 104.3, 90.7, 114.5, 120.6, 119.3, 121.3, 104.4, 92.2, 103.6, 99.6, 95.9, 114, 83.6, 110.6, 90.2, 98.3, 104.2, 108.3, 55.1, 106, 107.4, 89.8, 100, 90.3
is from a normal distribution.
y = c(120.8, 99, 104, 87.8, 121.7, 85.9, 104.3, 90.7, 114.5, 120.6, 119.3, 121.3, 104.4, 92.2, 103.6, 99.6, 95.9, 114, 83.6, 110.6, 90.2, 98.3, 104.2, 108.3, 55.1, 106, 107.4, 89.8, 100, 90.3)
# Anderson-Darling Normality Test, with parameters estimated
ad.test(y, "pnorm", estimated = TRUE) ##
## Anderson-Darling test of goodness-of-fit
## Braun's adjustment using 5 groups
## Null hypothesis: Normal distribution
## Parameters assumed to have been estimated from data
##
## data: y
## Anmax = Inf, p-value = 0.0004999# Anderson-Darling Normality Test, with parameters provided
ad.test(y, "pnorm", mean = 100, sd = 15) ##
## Anderson-Darling test of goodness-of-fit
## Null hypothesis: Normal distribution
## with parameters mean = 100, sd = 15
## Parameters assumed to be fixed
##
## data: y
## An = 0.76041, p-value = 0.5093shapiro.test(y) # Shapiro-Wilk Normality Test##
## Shapiro-Wilk normality test
##
## data: y
## W = 0.92312, p-value = 0.03234The first test rejects normality, while the other two don’t.
Chapter 10. Survival Analysis
Survival analysis is a branch of statistics and its primary purpose is to analyze time to event data. Examples of time to event include
time to death,
time until an electrical component fails,
time to recidivism,
time to first recurrence of a tumor after initial treatment, and
time to first marriage.
For more information,
Watch a introductory video: https://datatab.net/tutorial/survival-analysis
Read this article: https://www.karger.com/article/FullText/324758
Let \(X\) represent the time to an event. \(X\) is a random variable. The cumulative distribution function of \(X\) is denoted by \(F(x)=P(X\le x)\) and the survival function of \(X\) is defined as \(S(x)=P(X>x)\). If \(X\) represents the time (say in years) to first recurrence of a tumor after initial treatment, then \(S(5)\) represents the percent of patients whose length of remission is greater than 5 years. If \(X\) represents the time (say in months) to first marriage, then \(S(5)\) represents the percent of people who take more than 5 years before starting their first marriage.
Given a sample, how can we estimate \(S(x)\) for every \(x\)? A natural estimate is the fractions of values that are greater \(x\).
Example 1.
A random sample of 10 light bulbs are selected from a warehouse. The selected light bulbs are tested until failure or reaching a maximum of 2000 hours. Here are the life times of these light bulbs: 483, 449, 1920, 680, 1084, 206, 2000+, 2000+, 2000+, 1569, where “2000+” means more than 2000, which is called a right censored value.
Estimate
\(S(300)\)
\(S(500)\)
\(S(1500)\)
\(S(2200)\)
Solution.
Sort the data to get: 206, 449, 483, 680, 1084, 1596, 1920, 2000+, 2000+, 2000+.
Let \(X\) denote the life time of a random light bulb.
\(S(300) = P(X>300)\approx \frac{9}{10} =0.9\)
\(S(500) = P(X>500)\approx \frac{7}{10} =0.7\)
\(S(1500) = P(X>1500)\approx \frac{5}{10} =0.5\)
\(S(2200) = P(X>2200)\approx \frac{?}{10}\)
Special methods are needed for handling question (4), such as the methods introduced in https://sphweb.bumc.bu.edu/otlt/mph-modules/bs/bs704_survival/bs704_survival4.html
The Kaplan-Meier Curve
Read these two papers: https://www.real-statistics.com/survival-analysis/kaplan-meier-procedure/kaplan-meier-overview/ and https://towardsdatascience.com/kaplan-meier-curves-c5768e349479
Example 2.
During the period from 3/1/2022 to 7/1/2002, twelve new credit cards were sent to 12 randomly chosen customers.
| Customer | DateOfCardIssue | FirstDayOfCardUse | day | status |
|---|---|---|---|---|
| 1 | 2022-03-02 | 2022-03-08 | 6 | 1 |
| 2 | 2022-03-04 | 2022-03-11 | 7 | 1 |
| 3 | 2022-03-10 | 2022-03-18 | 8 | 1 |
| 4 | 2022-03-28 | 2022-04-16 | 19 | 1 |
| 5 | 2022-04-07 | 2022-04-23 | 16 | 1 |
| 6 | 2022-04-16 | 2022-05-02 | 16 | 1 |
| 7 | 2022-05-01 | 2022-05-14 | 13 | 1 |
| 8 | 2022-05-28 | NA | 34 | 0 |
| 9 | 2022-06-01 | 2022-06-13 | 12 | 1 |
| 10 | 2022-06-09 | 2022-06-24 | 15 | 1 |
| 11 | 2022-06-14 | NA | 17 | 0 |
| 12 | 2022-06-13 | NA | 18 | 0 |
| Note: | ||||
We will use the R package “survival” to create a Kaplan-Meier curve for the “survival” time defined to be the number of days it takes a customer to use the new credit card. Use the “Surv” function to specify “survival” time and status (1 = event happened, 0 = censored). Use the “survfit” function to calculate the survival probabilities. A reference: https://rviews.rstudio.com/2017/09/25/survival-analysis-with-r/
# Need to load package "survival"
fit <- survfit(Surv(day, status) ~ 1, # "1" means there is no explanatory variable
data = D,
conf.type = "plain", # There are 3 types: "plain", "log" (default), and "log-log"
conf.int = 0.95
)
autoplot(fit) # Need package "ggfortify"
summary(fit) # Produce a detailed table with estimate of survival probabilities and CI's## Call: survfit(formula = Surv(day, status) ~ 1, data = D, conf.type = "plain",
## conf.int = 0.95)
##
## time n.risk n.event survival std.err lower 95% CI upper 95% CI
## 6 12 1 0.917 0.0798 0.7603 1.000
## 7 11 1 0.833 0.1076 0.6225 1.000
## 8 10 1 0.750 0.1250 0.5050 0.995
## 12 9 1 0.667 0.1361 0.3999 0.933
## 13 8 1 0.583 0.1423 0.3044 0.862
## 15 7 1 0.500 0.1443 0.2171 0.783
## 16 6 2 0.333 0.1361 0.0666 0.600
## 19 2 1 0.167 0.1361 0.0000 0.433The standard errors in the summary table are calculated using the Greenwood formula as given in https://www.real-statistics.com/survival-analysis/kaplan-meier-procedure/confidence-interval-for-the-survival-function/.
\[se = \hat{S(t)}\sqrt{\sum_{j=1}^k\frac{d_j}{n_j(n_j-d_j)}}\]
More references:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3932959/
https://www.karger.com/article/FullText/324758 (More details)
Log-rank Test
To compare survival curves among a few groups, we use the log-rank test, which tests the following hypotheses:
\[H_0: \text{There is no difference in survival probability among the groups.}\]
\[H_a: \text{There is a difference in survival probability among the groups.}\]
If the p-value of the test is less than some significance level (e.g. \(\alpha= 0.05\)), then we can reject the null hypothesis and conclude that there is sufficient evidence that there is a difference in survival probability among the groups.
Here is a nice reference showing the detail of calculation of the test statistic for log-rank test: https://sphweb.bumc.bu.edu/otlt/mph-modules/bs/bs704_survival/BS704_Survival5.html#headingtaglink_3. Another more detailed reference is https://web.stanford.edu/~lutian/coursepdf/unitweek3.pdf. Try them out.
A note: The reference uses a simplified version of the formula used by R for calculating the test statistic. To get the result consistent with R, use the
\[\frac{(\sum d_1-\sum e_1)^2}{\sum \frac{d(n-d)}{n-1}\cdot \frac{n_1}{n}\cdot (1-\frac{n_1}{n})}\] where
\(d_1\) is the number of events from group 1,
\(e_1\) is the expected number of events from group 1,
\(d\) is the total number of events from both groups,
\(n\) is the number of individuals at risk (right before each event time), and
\(n_1\) is the number of individuals from group 1 that are at risk.
All of the quantities in the above formula are computed at each event time. The summations are taken over those times (or equivalently, those event times since \(d\) and \(e\) are both 0 at any censored time).
To perform a log rank test in R, we can use the “survdiff()” function from the “survival” package, as shown by the following example.
Example 3.
We use the “ovarian” dataset from the “survival” package. The dataset contains information about 26 patients, including:
The survival time (futime, in months) after being diagnosed with ovarian cancer
The censoring status (fustat, 1 = died, 0 = censored)
The type (rx = 1 or rx = 2) of treatment received
Here is part of the data:
D = ovarian # from the survival package
D = rbind(names(D), D)
names(D) = NULL
rownames(D) = NULL
kable(D, align='cccccc',
caption = "<center><strong>Ovarian Cancer Survival Data
</strong></center>",
table.attr = "style='width:60%; '") %>%
footnote("")%>%
kable_classic(full_width = TRUE, position = "center") %>%
column_spec (1:6,border_left = TRUE, border_right = TRUE) %>%
kable_styling("bordered") # Options: basic, striped, bordered, hover, condensed, responsive and none.| futime | fustat | age | resid.ds | rx | ecog.ps |
| 59 | 1 | 72.3315 | 2 | 1 | 1 |
| 115 | 1 | 74.4932 | 2 | 1 | 1 |
| 156 | 1 | 66.4658 | 2 | 1 | 2 |
| 421 | 0 | 53.3644 | 2 | 2 | 1 |
| 431 | 1 | 50.3397 | 2 | 1 | 1 |
| 448 | 0 | 56.4301 | 1 | 1 | 2 |
| 464 | 1 | 56.937 | 2 | 2 | 2 |
| 475 | 1 | 59.8548 | 2 | 2 | 2 |
| 477 | 0 | 64.1753 | 2 | 1 | 1 |
| 563 | 1 | 55.1781 | 1 | 2 | 2 |
| 638 | 1 | 56.7562 | 1 | 1 | 2 |
| 744 | 0 | 50.1096 | 1 | 2 | 1 |
| 769 | 0 | 59.6301 | 2 | 2 | 2 |
| 770 | 0 | 57.0521 | 2 | 2 | 1 |
| 803 | 0 | 39.2712 | 1 | 1 | 1 |
| 855 | 0 | 43.1233 | 1 | 1 | 2 |
| 1040 | 0 | 38.8932 | 2 | 1 | 2 |
| 1106 | 0 | 44.6 | 1 | 1 | 1 |
| 1129 | 0 | 53.9068 | 1 | 2 | 1 |
| 1206 | 0 | 44.2055 | 2 | 2 | 1 |
| 1227 | 0 | 59.589 | 1 | 2 | 2 |
| 268 | 1 | 74.5041 | 2 | 1 | 2 |
| 329 | 1 | 43.137 | 2 | 1 | 1 |
| 353 | 1 | 63.2192 | 1 | 2 | 2 |
| 365 | 1 | 64.4247 | 2 | 2 | 1 |
| 377 | 0 | 58.3096 | 1 | 2 | 1 |
| Note: | |||||
Plot the survival curve for each treatment group. Overlay the curves.
Test if there is any difference in survival probability between the two treatment groups.
Solution.
- Plot the survival curves.
# Plot and overlay survival curves: Need package "ggfortify"
autoplot(survfit(Surv(futime, fustat) ~ rx, data = ovarian, conf.type="plain"),
xlab = "Time",
ylab = "Overall survival probability")
The survival curves appear slightly different, but confidence bands overlap. Is the difference significant? We test it in part (b).
- Test whether there is a difference in survival probability between the two treatment groups.
fit = survdiff(Surv(futime, fustat) ~ rx,data=ovarian)
fit## Call:
## survdiff(formula = Surv(futime, fustat) ~ rx, data = ovarian)
##
## N Observed Expected (O-E)^2/E (O-E)^2/V
## rx=1 13 7 5.23 0.596 1.06
## rx=2 13 5 6.77 0.461 1.06
##
## Chisq= 1.1 on 1 degrees of freedom, p= 0.3Since the \(p\)-value is 0.3, greater than any commonly used significance level, the log rank test suggests no statistically significant difference between the two treatment groups.
Fit Cox Proportional Hazards Regression Model
The hazard function is defined as
\[h(t)=\frac{f(t)}{S(t)}\]
Where
- \(f(t)\) = the probability density function of survival time \(T\),
- \(S(t)\) = the Survival function (the probability of surviving beyond a certain point in time)
Cox proportional hazards regression model can be written as follows:
\[h(t)=h_0(t)e^{\beta_1\cdot x_1 + \beta_2\cdot x_2 +\cdots+ \beta_p\cdot x_p }\]
where
where h(t) is the expected hazard at time t, \(h_0(t)\) is the baseline hazard representing the hazard when all of the predictors (or independent variables) \(x_1, x_2, \cdots, x_p\) are equal to zero.
There are several important assumptions for appropriate use of the Cox proportional hazards regression model, including
independence of survival times between distinct individuals in the sample,
a multiplicative relationship between the predictors and the hazard, and
a constant hazard ratio over time.
The Cox proportional hazards model is called a semi-parametric model, because there are no assumptions about the shape of the baseline hazard function, except for other assumptions such as the three assumptions stated above.
Example 4 (Recidivism).
To demonstrate the Cox model, we follow the reference https://socialsciences.mcmaster.ca/jfox/Books/Companion/appendices/Appendix-Cox-Regression.pdf and use the “Rossi” data from the “carData” package. The data pertain to 432 convicts who were released from Maryland state prisons in the 1970s and who were followed up for one year after release. Half the released convicts were assigned at random to an experimental treatment in which they were given financial aid; half did not receive aid.
We use the following variables:
week: week of first arrest after release, or censoring time.
arrest: the event indicator, equal to 1 for those arrested during the period of the study and 0 for those who were not arrested.
fin: a factor, with levels “yes” if the individual received financial aid after release from prison, and “no” if he did not; financial aid was a randomly assigned factor manipulated by the researchers.
age: in years at the time of release.
race: a factor with levels “black” and “other”.
wexp: a factor with levels “yes” if the individual had full-time work experience prior to incarceration and “no” if he did not.
mar: a factor with levels “married” if the individual was married at the time of release and “not married” if he was not.
paro: a factor coded “yes” if the individual was released on parole and “no” if he was not.
prio: number of prior convictions.
educ: education, a categorical variable coded numerically, with codes 2 (grade 6 or less), 3 (grades 6 through 9), 4 (grades 10 and 11), 5 (grade 12), or 6 (some post-secondary).
emp1-emp52: factors coded “yes” if the individual was employed in the corresponding week of the study and “no” otherwise.
head(Rossi) # From package "carData"## week arrest fin age race wexp mar paro prio educ emp1 emp2 emp3 emp4
## 1 20 1 no 27 black no not married yes 3 3 no no no no
## 2 17 1 no 18 black no not married yes 8 4 no no no no
## 3 25 1 no 19 other yes not married yes 13 3 no no no no
## 4 52 0 yes 23 black yes married yes 1 5 no no no no
## 5 52 0 no 19 other yes not married yes 3 3 no no no no
## 6 52 0 no 24 black yes not married no 2 4 no no no no
## emp5 emp6 emp7 emp8 emp9 emp10 emp11 emp12 emp13 emp14 emp15 emp16 emp17
## 1 no no no no no no no no no no no no no
## 2 no no no no no yes yes yes yes yes no no no
## 3 no no no no no no no no no no no no yes
## 4 yes yes yes yes yes yes yes yes yes yes yes yes yes
## 5 no no no no no yes yes yes yes yes yes yes yes
## 6 yes yes yes yes yes yes no no no no no no no
## emp18 emp19 emp20 emp21 emp22 emp23 emp24 emp25 emp26 emp27 emp28 emp29 emp30
## 1 no no no <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA>
## 2 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA>
## 3 no no no no no no no no <NA> <NA> <NA> <NA> <NA>
## 4 yes yes yes yes no no no no no no no no no
## 5 yes yes yes yes yes yes yes yes yes yes yes yes yes
## 6 no no no no no yes yes yes yes yes yes yes no
## emp31 emp32 emp33 emp34 emp35 emp36 emp37 emp38 emp39 emp40 emp41 emp42 emp43
## 1 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA>
## 2 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA>
## 3 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA>
## 4 no yes yes yes yes yes yes yes yes yes yes yes yes
## 5 yes yes yes yes no no no no no no no no no
## 6 no no no no no no no no no no no no no
## emp44 emp45 emp46 emp47 emp48 emp49 emp50 emp51 emp52
## 1 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA>
## 2 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA>
## 3 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA>
## 4 yes yes yes yes yes yes yes yes yes
## 5 no no no no no no no no no
## 6 no no no no no no no no nofit <- coxph(Surv(week, arrest) ~ fin + age + race + wexp + mar + paro + prio, data=Rossi)
summary(fit)## Call:
## coxph(formula = Surv(week, arrest) ~ fin + age + race + wexp +
## mar + paro + prio, data = Rossi)
##
## n= 432, number of events= 114
##
## coef exp(coef) se(coef) z Pr(>|z|)
## finyes -0.37942 0.68426 0.19138 -1.983 0.04742 *
## age -0.05744 0.94418 0.02200 -2.611 0.00903 **
## raceother -0.31390 0.73059 0.30799 -1.019 0.30812
## wexpyes -0.14980 0.86088 0.21222 -0.706 0.48029
## marnot married 0.43370 1.54296 0.38187 1.136 0.25606
## paroyes -0.08487 0.91863 0.19576 -0.434 0.66461
## prio 0.09150 1.09581 0.02865 3.194 0.00140 **
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## exp(coef) exp(-coef) lower .95 upper .95
## finyes 0.6843 1.4614 0.4702 0.9957
## age 0.9442 1.0591 0.9043 0.9858
## raceother 0.7306 1.3688 0.3995 1.3361
## wexpyes 0.8609 1.1616 0.5679 1.3049
## marnot married 1.5430 0.6481 0.7300 3.2614
## paroyes 0.9186 1.0886 0.6259 1.3482
## prio 1.0958 0.9126 1.0360 1.1591
##
## Concordance= 0.64 (se = 0.027 )
## Likelihood ratio test= 33.27 on 7 df, p=2e-05
## Wald test = 32.11 on 7 df, p=4e-05
## Score (logrank) test = 33.53 on 7 df, p=2e-05Interpretation of the results:
The exponentiated coefficients in the second column of the first panel (and in the first column of the second panel) of the output are interpretable as multiplicative effects on the hazard. Thus, for example,
holding the other covariates constant, an additional year of age reduces the weekly hazard of rearrest by a factor of 0.94418 on average, or by about 5.6%.
Similarly, each prior conviction increases the hazard by a factor of 1.096, or 9.6 percent.
The likelihood-ratio, Wald, and score chi-square statistics at the bottom of the output are asymptotically equivalent tests of the omnibus null hypothesis that all of the \(\beta\)’s are zero. In this instance, the test statistics are in close agreement, and the omnibus null hypothesis is soundly rejected.
We may wish to display how estimated survival probability depends upon the value of a covariate. Because the principal purpose of the recidivism study was to assess the impact of financial aid on rearrest, we focus on this covariate. We construct a new data frame with two rows, one for each value of “fin”; the other covariates are fixed to their average values. For a dummy covariate, such as the contrast associated with “race”, the average value is the proportion coded 1 in the data set (in the case of race, the proportion of non-blacks).
Rossi.fin <- data.frame(fin=c("no", "yes"),
age=mean(Rossi$age),
race=mean(Rossi$race == "other"),
wexp=mean(Rossi$wexp == "yes"),
mar=mean(Rossi$mar == "not married"),
paro=mean(Rossi$paro == "yes"),
prio=mean(Rossi$prio)
)
Rossi.fin## fin age race wexp mar paro prio
## 1 no 24.59722 0.1226852 0.5717593 0.8773148 0.6180556 2.983796
## 2 yes 24.59722 0.1226852 0.5717593 0.8773148 0.6180556 2.983796par(mgp=c(1.5,0.5,0), # The margin line (in mex units) for the axis title,
# axis labels and axis line. Note that mgp[1] affects
# title whereas mgp[2:3] affect axis. The default is c(3, 1, 0).
mar = c(5,4,1,3)+0.1 # A numerical vector of the form c(bottom, left, top, right) # which gives the number of lines of margin to be specified on # the four sides of the plot.
# The default is c(5, 4, 4, 2) + 0.1.
)
plot(survfit(fit, newdata=Rossi.fin),
conf.int=TRUE,
lty=c(1, 2),
ylim=c(0.65, 1),
xlab="Weeks",
ylab="Proportion Not Rearrested",
cex.lab = 0.8,
cex.axis = 0.8,
col = c(1,2))
legend("bottomleft",
legend=c("fin = no", "fin = yes"),
lty=c(1, 2),
inset=0.02,
bty = "n",
col = c(1,2))
text<- "Figure 3: Estimated survival functions for those receiving (fin = yes) and not receiving (fin = no) \nfinancial aid. Other covariates are fixed at their average values. Each estimate is accompanied \nby a point-wise 95-percent confidence envelope."
mtext(text, side = 1, line = 4, cex = 0.8, adj = 0)
The resulting graph shows the higher estimated “survival” of those receiving financial aid, but the two confidence envelopes overlap substantially, even after 52 weeks.
References
1a. https://faculty.ksu.edu.sa/sites/default/files/nonparametric_statistics_a_step-by-step_approach.pdf
1b. A classic: http://www.ru.ac.bd/wp-content/uploads/sites/25/2019/03/303_11_Siegel-Nonparametric-statistics-for-the-behavioral-sciences.pdf
Compare two unrelated samples using t-Test for independent samples: The corresponding non-parametric counterpart is: Mann–Whitney U-test and Kolmogorov–Smirnov two-sample test; permutation test (or randomization test): https://www.jwilber.me/permutationtest/ and https://thomasleeper.com/Rcourse/Tutorials/permutationtests.html
Compare two related samples using t-Test for dependent samples: The corresponding non-parametric counterpart is: Wilcoxon signed ranks test and sign test
Compare three or more unrelated samples using One-way ANOVA: The corresponding non-parametric counterpart is: Kruskal–Wallis H-test
Compare three or more related samples using repeated measures ANOVA: https://statistics.laerd.com/statistical-guides/repeated-measures-anova-statistical-guide.php
The corresponding non-parametric counterpart is the Friedman test and Kendall test:
Compare two quantitative variables using Pearson product–moment correlation: The corresponding non-parametric counterpart is: Spearman rank-order correlation method https://www.statisticshowto.com/probability-and-statistics/correlation-coefficient-formula/spearman-rank-correlation-definition-calculate/, Point-biserial correlation, or Biserial correlation
How could you write R code to do it?
Runs test when examining a sample for randomness
Skip!
Chi square tests and Fisher exact test when comparing two categorical variables
Survival data analysis: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3932959/ https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3059453/ (censored is indicated by a tick mark but no jump in plots)
Rater Data: https://www.usu.edu/math/jrstevens/biostat/projects2013/pres_mcnemarcohen.pdf
Cohen’s Kappa and inter-rater agreement: https://www.youtube.com/watch?v=djd7GalIB8c McNemar’s test for symmetry (2 categories): <> McNemar-Bowker test for symmetry (more than 2 categories): https://ncss-wpengine.netdna-ssl.com/wp-content/themes/ncss/pdf/Procedures/PASS/Tests_for_Multiple_Correlated_Proportions-McNemar-Bowker_Test_of_Symmetry.pdf
https://www.youtube.com/watch?v=KmOw3a_4RmI (Using excel) Base R function: mcnemar.test()
- Power and sample size determination: https://sphweb.bumc.bu.edu/otlt/mph-modules/bs/bs704_power/bs704_power_print.html
Mathematical Foundation to Non-Parametric Statistics
Given a sample of size n, at most how many bootstrap samples are possible? That is, how many ways are there when sampling k items from a set of n distinct ones? Here k = n. A special stick-stone problem. When n = 3, list all possible bootstrap samples.
Readings
A Comparison of Parametric and Non-Parametric Methods Applied to a Likert Scale: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5597151/
Appendix
More than one individual receives a treatment in each block of an RCBD
Only consider the situation where both block and treatment are fixed.
Block=rep(c("B1", "B2", "B3"),c(9,9,9))
Treatment=rep(rep(c("T1", "T2", "T3"),c(3,3,3)),3)
y = c(2,1,3,4,2,2,3,4,4,4,5,4,4,5,6,5,6,7,6,7,6,7,8,7,9,8,7)
D=data.frame(Block,Treatment,y)
m = aov(y~Treatment+Error(Block/Treatment))
summary(m)##
## Error: Block
## Df Sum Sq Mean Sq F value Pr(>F)
## Residuals 2 88.96 44.48
##
## Error: Block:Treatment
## Df Sum Sq Mean Sq F value Pr(>F)
## Treatment 2 12.519 6.259 169 0.000137 ***
## Residuals 4 0.148 0.037
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Error: Within
## Df Sum Sq Mean Sq F value Pr(>F)
## Residuals 18 13.33 0.7407# Alternatively, use a regression approach with interaction
m= lm(y~Block+Treatment+Treatment:Block)
anova(m)## Analysis of Variance Table
##
## Response: y
## Df Sum Sq Mean Sq F value Pr(>F)
## Block 2 88.963 44.481 60.05 1.086e-08 ***
## Treatment 2 12.519 6.259 8.45 0.002582 **
## Block:Treatment 4 0.148 0.037 0.05 0.994875
## Residuals 18 13.333 0.741
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1The Power Function of a Test
For any test of hypotheses, there are two types of error:
Type I error. This occurs when the null hypothesis is true but is rejected.
Type II error. This occurs when the null hypothesis is false but is not rejected.
The probability of making type I error is denoted by \(\alpha\). The probability of making type II error is denoted by \(\beta\). Thus, the probability of correctly rejecting a false null hypothesis equals \(1-\beta\), which is called the power of the test.
Here is a tutorial about the calculation of the power for a one-sample t-test: https://stats.oarc.ucla.edu/r/dae/power-analysis-for-one-sample-t-test/
pwr.t.test(d=(95 - 100)/15,
n=85,
sig.level=0.05,
type="one.sample",
alternative="less")##
## One-sample t test power calculation
##
## n = 85
## d = -0.3333333
## sig.level = 0.05
## power = 0.9197555
## alternative = lessWhen the effect size is -1/3, for a sample size of 85, the power is about 0.92 to reject \(H_0:\mu=100\) under the alternative hypothesis \(H_a:\mu=95\).
Next, suppose we want a power 0.80, what sample size is needed keeping all of the other numbers the same?
pwr.t.test(d=(95 - 100)/15,
power = 0.80,
sig.level=0.05,
type="one.sample",
alternative="less")##
## One-sample t test power calculation
##
## n = 57.02048
## d = -0.3333333
## sig.level = 0.05
## power = 0.8
## alternative = lessThe result tells us that, when effect size equals -1/3, we need a sample size at least 57 in order to have a power of 0.80 to reject \(H_0:\mu=100\) under the alternative hypothesis \(H_a:\mu=95\).
Check your calculation using the online power calculator: https://www.stat.ubc.ca/~rollin/stats/ssize/n1.html