Hw2_Group_1
Homework 1 - to be done as groups
Names: Elisa, Emilie, Lív, Victor, Viktoria and Weizhe
Group: 1
For deadlines etc, see Absalon.
You have to supply both the answer (numbers, a table, plots or combinations thereof) as well as the R or Linux code you used to make the plots. This should be done using this R markdown template: we want both the R markdown file and a resulting PDF. For PDF output, you may have to install some extra programs - RStudio will tell you.
Note that:
If the R code gives different results than your results, you will get severe point reductions or even 0 points for the exercise
Some questions may request you to use R options we have not covered explicitly in the course; this is part of the challenge
While this is a group work, we expect that everyone in the group will have understood the group solution: similar or harder question might show up in the individual homework. So, if something is hard, it means you need to spend more time on it
The results should be presented on a level of detail that someone else could replicate the analysis.
For statistical tests, you have to:
Motivate the choice of test
State exactly what the null hypothesis is (depends on test!)
Comment the outcome: do you reject the null hypothesis or not, and what does this mean for the actual question we wanted to answer (interpretation)?
A question marked * means that is more challenging, and likely requires skills from the whole group.
Question 1: Dicer dissected
The human DICER1 gene encodes an important ribonuclease, involved in miRNA and siRNA processing. Several mRNAs representing this gene have been mapped to the human genome (March 2006 assembly). We will look closer at one of them with the accession number AK002007.
- What are the first five genomic nucleotides that are read by RNA polymerae II from this transcript?
When looking up the accession number AK002007, we see that this
particular mapping is from the “Human mRNA from GenBank” and is
positioned at chromosome 14: 94626558-94636029. 
When we open the alignment, wee see that the first 5 genomic
nucleotides read by RNApol-II is AAAGG. 
- Look at the raw mRNA sequence of AK002007, from the database it
actually comes from. What are the first five nucleotides?

As seen in both the picture of the raw mRNA-sequence above and the alignment-picture, the first five nucleotides from the transcribed sequence and the raw mRNA are different. Here, the five first nucleotides are GAAGC
- How do you explain the discrepancy (maximum 5 lines)?
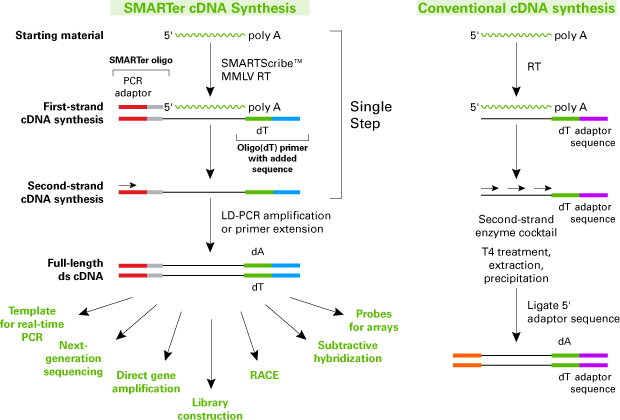
As there is discrepancy between the raw and the cDNA sequence, there is a possibility that those “extra” nucleotides are “left-over” from the preparation of the sample. The below picture shows the mapping of this sequence to the genbank, and we cannot see the additional 7 nucleotides. This argues that they hold no genomic and/or biological significance. As we do not know the exact methods behind finding the sequence, we can propose that they are an adaptor-sequence, meaning an short oligonucleotide added to aid the cDNA synthesis. Source: https://www.takarabio.com/images/Data%20Image/1231.gif
{kind=link}

Q1.c
Question 2: ERA and ERB
Our collaborators designed a ChIP study using so-called tilling arrays (an outdated technique these days, but the top of the pop at the time: see http://en.wikipedia.org/wiki/Tiling_array): one for estrogen receptor alpha (ERA), one for estrogen receptor beta (ERB). All the sites are stored in BED files respectively for two ERs. These are now available in the homework directory, and are both mapped on hg18 genome. The current situation is that we know to some degree what ERA does, but not what ERB does (there are some evidence that they share some functions, but not all). So, we need bigger experiments and better statistics.
- Using BEDtools within Linux: What is the genome coverage (% of base pair covered at each chromosome) for ERB and ERA sites? If you need a file with chromosome sizes for hg18, it included in the assignment: hg18_chrom_sizes.txt. Plot the fractions for all chromosomes as a single barplot in R. Briefly comment the results. Is there anything particularly surprising? Try to explain the outcome (biological and/or experimental setup explanations)?
I upload the three files: “ERa_hg18.bed”, “ERb_hg18.bed” and “hg18_chrom_sizes.txt” to a newly created wd within the Linux server. I then take a look at them, to figure out what they contain. To merge and map files ontop of each other, it is important that they are sorted first. This is done by the “sort”-function.
Unix commands…
sort -k1,1 -k2,2n ERa_hg18.bed > sorted_ERa_hg18.bed
sort -k1,1 -k2,2n ERb_hg18.bed > sorted_ERb_hg18.bed
…making two new bed files that are sorted. These can now be used to map to the genome, which here is “hg18_chrom_sizes.txt”
Unix commands:
nice bedtools genomecov -i sorted_ERa_hg18.bed -g hg18_chrom_sizes.txt -max 1 > cov_ERa_hg18.bed
nice bedtools genomecov -i sorted_ERb_hg18.bed -g hg18_chrom_sizes.txt -max 1 > cov_ERb_hg18.bed
ERa_cov <- read_tsv("cov_ERa_hg18.bed", col_names = c("chr", "overlap", "no_bases", "chr_size", "fraction"))## Rows: 33 Columns: 5
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: "\t"
## chr (1): chr
## dbl (4): overlap, no_bases, chr_size, fraction
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.ERb_cov <- read_tsv("cov_ERb_hg18.bed", col_names = c("chr", "overlap", "no_bases", "chr_size", "fraction"))## Rows: 33 Columns: 5
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: "\t"
## chr (1): chr
## dbl (4): overlap, no_bases, chr_size, fraction
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.ER_cov <- bind_rows("ERa" = ERa_cov, "ERb" = ERb_cov, .id = "type") %>% filter(chr != "genome", chr != "chrM")
ER_cov %>%
filter(overlap == 0) %>%
ggplot(aes(x = chr, y = 1- fraction, fill = type))+
geom_bar(stat = "identity", position = "dodge")+
xlab("Chromosome")+
ylab("Coverage Fraction")+
ggtitle("Fraction of Coverage for All Chromosomes")+
theme_light()+
theme(axis.text.x = element_text(angle = 75, hjust = 1.2))
We see from the plot that both receptors bind to the same chromosomes and that the ERa has a higher chromosome coverage than ERb; E.g. there are twice as many biding sites for ERa on chromosome X as there are for ERb. There are both be a biological explanation as well as a methodological explanation for this.
Receptors bind to the same chromosome, but that does not mean that they regulate the same genes necessarily, but there might be redundancy.
They probably have similar functions, e.g. regulate the same genes or neighbouring genes.
Victor’s comments
- Again, using BEDtools in Linux: How many ERA sites do/do not overlap ERB sites, and vice versa? Show the Linux commands and then a Venn diagram summarizing the results. The Venn diagram can be made in R using one of many venn diagram packages, but you can also make it in any drawing program.
Unix commands:
We first have to clean the data, which is done by the way Ellen
suggested on Absalon.
Then we run the following codes:
bedtools intersect -a ERa_hg18.bed -b ERb_hg18.bed -c > ERa_on_ERb.bed
bedtools intersect -a ERb_hg18.bed -b ERa_hg18.bed -c > ERb_on_ERa.bed
^For ease, I have just written the former names of the raw files, but in reality I saved the cleaned files as new .bed-files.
library(VennDiagram)## Indlæser krævet pakke: grid## Indlæser krævet pakke: futile.loggerERa_ERb <- read_tsv("ERa_on_ERb.bed", col_names = c("chr", "start", "end","overlap"))## Rows: 581 Columns: 4## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: "\t"
## chr (1): chr
## dbl (3): start, end, overlap
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.ERb_ERa <- read_tsv("ERb_on_ERa.bed", col_names = c("chr", "start", "end","overlap"))## Rows: 485 Columns: 4
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: "\t"
## chr (1): chr
## dbl (3): start, end, overlap
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.overlap <- ERa_ERb %>% filter(overlap == 1) %>% count()
ERa <- ERa_ERb %>% count()
ERb <- ERb_ERa %>% count()
venn <- draw.pairwise.venn(area1 = ERa$n, area2 = ERb$n, cross.area = overlap$n,
category = c("ERa", "ERb"),
lty = rep("blank", 2),
fill = c("light blue", "pink"),
alpha = rep(0.5, 2),
cat.pos = c(0, 0),
cat.dist = rep(0.025, 2))
Victor’s comments.
Question 3: Ribosomal Gene (*)
Your group just got this email from a frustrated fellow student:
My supervisor has found something he thinks is a new ribosomal protein gene in mouse. It is at chr9:24,851,809-24,851,889, assembly mm8. His arguments for this are:
a) It has high conservation in other species because ribosomal protein genes from other species map to this mouse region
b) And they are all called Rpl41 in the other species (if you turn on the other Refseq you see this clearly in fly and other species).
But, I found out that if you take the fly refseq sequence mentioned above (from Genbank) and BLAT this to the fly genome, you actually get something that looks quite different from the one in the mouse genome. How can this be? Is the mouse gene likely to be real? If not, why? (Maximum 20 lines, plus possibly genome browser pictures)