class: center, middle, inverse, title-slide # Differential Gene Expression Analysis of Fulvestrant-Treated MCF-7 RNA-seq Data ## Processed and Analyzed with RSubread and EdgeR ### Robert Bartolini ### University of Massachusetts Boston ### 2020/12/22 --- ```r #Rsubread builds reference genome index, maps sequence reads to annotated index library(Rsubread) #For DGE analysis and visualization library(edgeR) #ggplot2 library(tidyverse) #interactive MD plot w/ gene-wise expression library(Glimma) #top gene table library(reactable) #annotation of top gene table library(annotate) library(org.Hs.eg.db) ``` --- ```r ####The purpose of the workflow is to find differentially expressed genes ##from a RNA-seq data of Estrogen-receptor+ breast cancer cell lines #(MCF-7), a group treated with #fulvestrant (estrogen receptor antagonist) and a vehicle group. #Data obtained from: #https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136823 #courtesy of:Zeynep Madak-Erdogan @ University of Illinois at Urbana #data was downloaded from Sequence Read Archive via #Amazon Web Services S3 could system #build list of the raw sequence reads (data obtained from SRA) #source: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136823 #credit: Zeynep Madak-Erdogan fastq.files <- list.files(path = "rnaseq/", pattern = ".fastq$", full.names = TRUE) ##abbreviate sample names fastq.abbrv <- substr(fastq.files, start = 8, stop = 9) ``` --- ```r ############################################################# ####### __ ___ ____ _ _ ___ _ _ ____ ####### ####### \ \ / / \ | _ \| \ | |_ _| \ | |/ ___| ####### ####### \ \ /\ / / _ \ | |_) | \| || || \| | | _ ####### ####### \ V V / ___ \| _ <| |\ || || |\ | |_| | ####### ####### \_/\_/_/ \_\_| \_\_| \_|___|_| \_|\____| ####### #Index Building & Gene Mapping is Computationally Intensive!! ###TAKES HOURS @ 8GB/RAM ###Enter at your own RISK!!ლ(ಠ益ಠლ) #build the index - this takes the entire human genome, decompresses it #and builds a hash table of key-value pairs of 16-bp sequences (keys) #are assigned to chromosomal locations (values) #this will be used to map the rna-seq reads to reference genome from: #GENCODE database via the following links: #ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_28/GRCh38.primary_assembly.genome.fa.gz # #index is in directory buildindex(basename="Ebi_Homo_Sapien_Index",reference="GRCh38.primary_assembly.genome.fa.gz", gappedIndex = TRUE, indexSplit = TRUE) ##################Rsubread Alignment################ ##### align() creates an index hash table #that mediates the mapping of sequence reads to the reference index align.stat <- align(index = "Ebi_Homo_Sapien_Index", readfile1 = fastq.files) #Output is .BAM file this is used for #normalizing and counting the sequence reads ``` --- ```r #build a vector of themapped read file names bam.files <- list.files(path = "rnaseq/", pattern = ".BAM$", full.names = TRUE) #featureCounts generates a matrix of read counts of mapped genes in each sample #strandSpecific = 2 because 'TruSeq Stranded mRNAseq Sample Prep kit' (Illumina). #Read 1 aligns to the ANTISENSE strand and Read 2 aligns to the SENSE strand, #this maps the mrna sequence to the annotated exons within inbuilt annotation fcRS <- featureCounts(bam.files, annot.inbuilt="hg38", strandSpecific = 2) ``` --- ```r ###This chunk is a files that were written for downstream processing or analysis ####write counts to table fcRS_counts <- fcRS$counts #output a txt file of count data write.table(x=data.frame(fcRS$annotation[,c("GeneID","Length")],fcRS$counts,stringsAsFactors=FALSE),file="RScountsEBI.txt",quote=FALSE,sep="\t",row.names=FALSE) #For edgeR quasi-likelihood DGE write.table(x=data.frame(fcRS$annotation[,c("GeneID")],fcRS$counts,stringsAsFactors=FALSE),file="counts.txt",quote=FALSE,sep="\t",row.names=FALSE) #load data into a table (for count distribution) EBI_RStrandedCountTable <- read.table(file = 'RScountsEBI.txt', header = TRUE) #load data into a table for quasi-likelihood DGE CountTable <- read.table(file = 'counts.txt', header = TRUE) #IMAGE NEXT SLIDE ``` --- <!-- --> --- ```r #######HISTOGRAM OF Fulvestrant Sample 1 gene expression v. # of genes ggplot(EBI_RStrandedCountTable) + geom_histogram(aes(x = F1_CAAGCTAG.ACATAGCG_L002_R1_001..1..fastq.subread.BAM), stat = "bin", bins = 250, colour = "lightblue", fill = "orange") + xlab("Raw expression counts") + ylab("Number of genes")+ xlim(0,20000)+ ylim(0,1760)+ theme_dark() ###PLOT ON NEXT SLIDE ``` --- <!-- --> --- ```r #scatter-plot to display mean-variance relationship ####write counts to table (needed for mean/variance dependence plot and #later use in contrast matrix) fcRS_counts <- fcRS$counts #generate a vector of gene-wise mean expression mean <- apply(fcRS_counts[,1:6], 1, mean) #The second argument '1' of 'apply' function indicates the function being applied to rows. Use '2' if applied to columns #Generate a vector of gene-wise variance variance <- apply(fcRS_counts[,1:6], 1, var) #assemble mean and variance into a dataframe MeanVariancedf <- data.frame(mean, variance) #scatter-plot to display mean-variance relationship ggplot(MeanVariancedf) + geom_point(aes(x=mean, y=variance), colour = "orange", alpha = .8) + scale_y_log10(limits = c(1,1e9)) + scale_x_log10(limits = c(1,1e6)) + geom_abline(intercept = 0, slope = 1, color="green")+ xlab("Mean Gene Expression")+ ylab("Variance of Gene Expression")+ theme_dark() ``` --- <!-- --> --- ```r #Prepare for PCA Plot #Create a DGEList object from featureCount object for downstream analysis DGEfulvestrant <- DGEList(counts=fcRS$counts, genes=fcRS$annotation[,c("GeneID","Length")]) ####Filter to keep genes which had more than 10 reads per million mapped #reads in at least two libraries. FilteredGenes <- rowSums(cpm(DGEfulvestrant) > 10) >= 2 #write to dataframe DGEfulvestrant <- DGEfulvestrant[FilteredGenes,] ##Normalization. Perform voom normalization: Adjusts for between sample #sequence library size so statisical analysis can be performed Normalization <- voom(DGEfulvestrant,designmatrix,plot=T) ``` --- ```r ####MULTI-DIMENSIONAL SCALING PLOT #### #distances on the plot approximate the typical #log2 fold changes between the samples. #This function is a variation on the usual multdimensional #scaling (or principle coordinate) plot, in that a distance #measure particularly appropriate for the microarray context #is used. The distance between each pair of samples #(columns) is the root-mean-square deviation (Euclidean distance) #for the top genes. Distances on the plot can be #interpreted as leading log2-fold-change, meaning the #typical (root-mean-square) log2-fold-change between the samples for the genes that distinguish those samples. #tidy sample names(needed for Multi-Dimensional (PCA) plot labels later on) fcRS$targets <- substr(fcRS$targets, start = 1, stop = 1) plotMDS(Normalization, labels = samples, xlim=c(-1,1),ylim=c(-1.0,.5)) ``` --- <!-- --> --- ```r #create shrinkage of dispersion and plot the biological coefficient #variation #create group factors for contrast group <- factor(c(1,1,1,2,2,2)) #DGElist creates a list with grouping factors for sample y <- DGEList(counts=fcRS_counts,group=group) #this filters lowly expressed genes with may increase the FDR - #debatable if the genes are important and requires post hoc analysis keep <- filterByExpr(y) #rewrites with genes expression counts > 10 y <- y[keep,,keep.lib.sizes=FALSE] #trimmed mean of M values(TMM) normalization allows comparison of different sized libraries #the libraries y <- calcNormFactors(y) #create design contrast for glm model design <- model.matrix(~group) #estimate dispersion using linear combination of gene and sample wise likelihood y <- estimateDisp(y,design) ``` --- ```r #Plot the Biological Coefficient of Variation vs log2CountperMillion Abundance plotBCV(y) ``` <!-- --> --- ```r #Fit a quasi-NB model with design matrix fit <- glmQLFit(y,design) ``` <!-- --> --- ```r #Plot shrinkage of counts plotQLDisp(fit) ``` <!-- --> --- ```r #plot log CPM vs. log-fold change with p-value #color scale to show magnitude of differential #gene expression plotMD(qlf, main = "Fulvestrant v. Vehicle") ``` <!-- --> --- ```r #F-test of fit qlf <- glmQLFTest(fit,coef=2) #topTags extracts the most statistically DEGs TopGENES <- topTags(qlf) #extract table of DEGS from s3 object TopTable <- TopGENES[["table"]] #use library(annotate) to include gene symbols gene_symbols <- getSYMBOL(c(rownames), data = 'org.Hs.eg.db') #bind symbols with top DEgenes ToptableSym <- cbind(gene_symbols, TopTable) #display in a table reactable(ToptableSym) #table on the next slide ``` --- 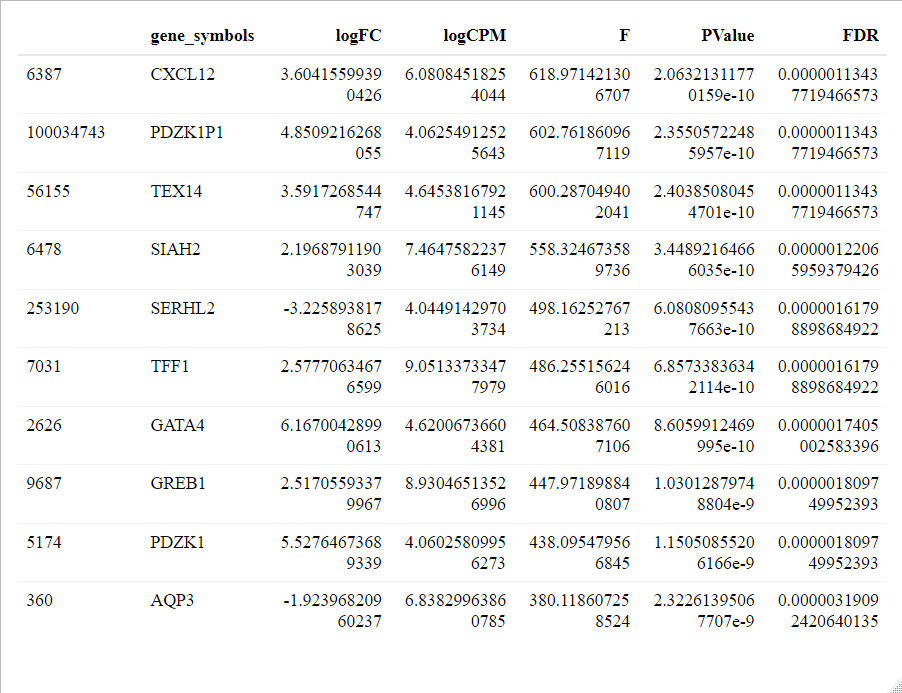<!-- -->