Deep Learning with R using TensorFlow via Keras
Leon Eyrich Jessen, PhD
August 21st 2018

Postdoctoral Researcher
Immunoinformatics and Machine Learning group
Department of Bio and Health Informatics
Technical University of Denmark
Talk
Department of Food Science
University of Copenhagen
 @jessenleon
@jessenleon
Who am I?

- My name is Leon Eyrich Jessen
- I have a background in wet lab biotech engineering (Think pipettes, goggles and lab coats)
- and a PhD in Bioinformatics (I will get back to what that is)
- I am a currently a Postdoctoral Researcher (What you do after your PhD, when you're too scared of the real world)
- I am in the Immunoinformatics and Machine Learning Group at the Technical University of Denmark
- I teach Introductory Data Science, Immunological Bioinformatics and I do guest lectures and workshops on deep learning at university at master level
So you do Bioinformatics - What is that?
- Bioinformatics is Data Science with domain specific knowledge in Biology

...and more specifically, I actually do Immunoinformatics
- I use neural networks to model molecular interactions in the human immune system in context of infectious diseases and cancer

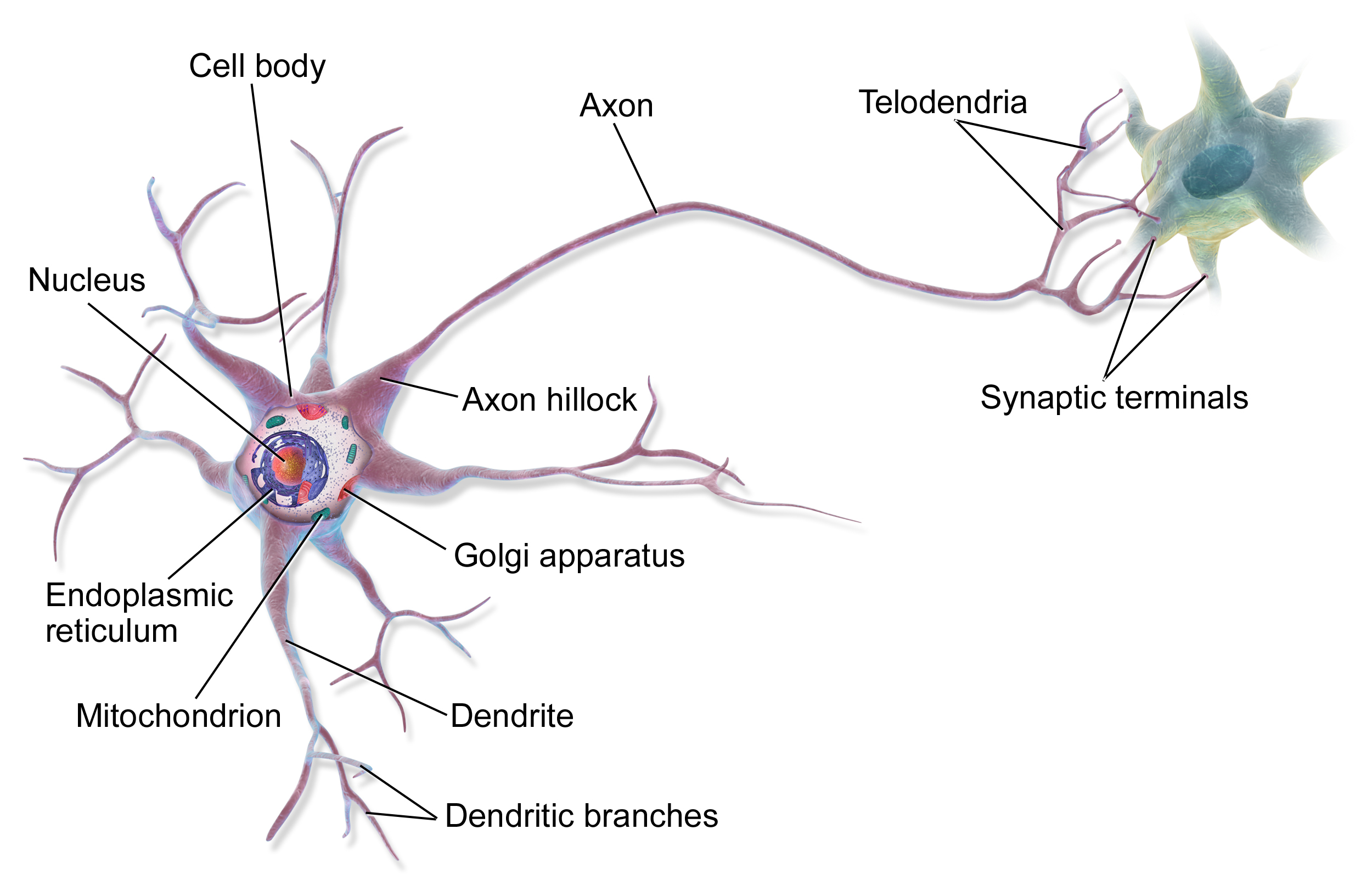
What are Artificial Neural Networks?

Source: Bruce Blaus | Multipolar Neuron | CC BY 3.0
{kind=link}
Basic Feed Forward Multilayer Perceptron - Architecture
- Fully connected dense neural network wih one input layer, one hidden layer and one output neuron

- Based on input, the neuron has to 'decide' whether to send a signal via the axon or not
Source: Bruce Blaus | Multipolar Neuron | CC BY 3.0
Why are artificial neural networs so powerful?
- ANNs can identify context dependence!
- I.e. the “meaning” of each variable depends not only on its observed value, but also on the context in which it was observed!
- I bet you can read this word quite easily, but take a closer look

Keras and TensorFlow - What you need to know
Let us get to it - Installing Keras and TensorFlow
Installing Keras and TensorFlow
A Couple of Examples
Example 1: Three-Class Classifier using the Classic Iris data set and Keras/TensorFlow
The Iris data set

The Iris data set

Neural Network Classifier Model
Here is an illustration of the model we will be building with 35 parameters, i.e. \( (n_{features} + 1) \cdot m_{hidden} + (m_{hidden} + 1) \cdot l_{output} \)

Prepare data
Scaling
Feature distributions before scaling
Feature distributions after scaling

Prepare data
Set Architecture, Compile Model and Train Classifier
Visualise training
The fit() function returns a history object, which we can plot like so
plot(history)

Evaluate Model Performance
Predict on New Data
Confusion Matrix Visualisation
…and finally we can visualise the confusion matrix based on the original 20% left out test data

Example 2: Modelling Molecular Interactions
Very Brief Background

Figure by Eric A. J. Reits
Cancer Immunotherapy

Source: Simon Caulton | Adoptive T-cell therapy | CC BY 3.0
{kind=link}
Data
Data
After encoding, each peptide is in essense converted to an image, i.e. a numeric representation of the biochemical profile of the peptide
pep_plot_encoding(pep = 'LLTDAQRIV')

Data
About Cross Validation

Defining and Compiling the Model
Training the model
Add Predictions to the Peptide Data Set
Visualisation of Predictions
Only allow each model to predict on the unseen test partition

Training
- Despite us only running the training for 10 epochs, the MSE decreases very rapidly!

Individual Model Performances
- The area under the Receiver Operator Characterstics curve (the ROC curve) is the so called AUC value.
- If \( AUC = 1 \), then you have a perfect predictor, if \( AUC = 0.5 \), then you have a random predictor

Wisdom of the crowd

Performance of overall model
Receiver Operator Characterstics curve

Performance on Left Out Test Set (Partition 6)
- For this set, we make 20 predictions for each \( x \) and assign the mean as the final prediction \( \hat{y} \)
Receiver Operator Characterstics curve
