One of the first algorithms I ever implemented computed the longest common subsequence. Apart from the imperative-heavy approach I used the code was reasonably good, but unfortunatly I lost it (along with many other scripts) in a recent hand-drive failure.
To celebrate my 1st anniverary of R usage I'm going to reimplement the algorithm, and run it over some sequence data to construct a phylogenetic tree.
Recursive definitions do not mesh well with R; they perform extremely poorly compared to their iterative counterparts. To counteract this you can use a technique called memoisation to speed up the code substantially. Simply put, memoisation is a technique in which you store the results of each function call, and if possible you use these stored results next time the same function call is used.
My implementation is slightly awkward; I created a recursive function, which calls the memoisation function. If the result has already been computed I return that, otherwise I calculate and store the new values. Still, it performs nicely compared to the naive recursive definition.
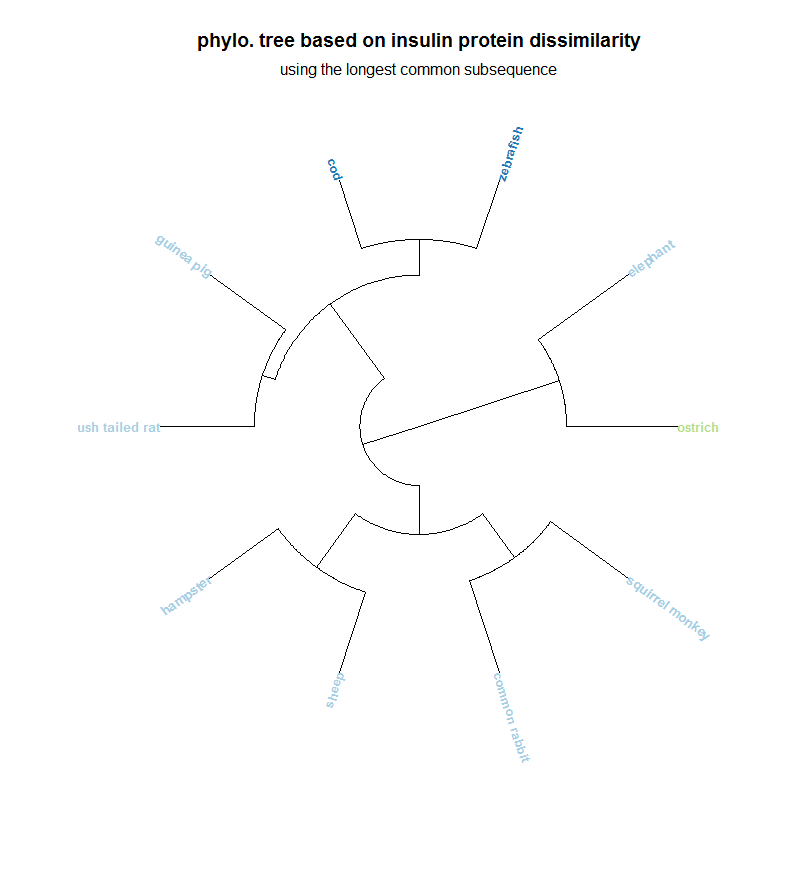
Please, do not regard this phylogenetic tree as being anyway accurate. Phylogenetic tree's aren't constructed with such a simple metric. If determining evolutionary relationships was as easy as using a bad metric on a tiny protein, it would have been done years ago. This was just meant to be a proof of concept, and a test case for the LCS function.
l <- base::length
LCS <- function(X, Y) {
if (prod(l(X), l(Y)) == 0)
return(list(sequence = c(), length = 0))
sequence = LCS_recursive(X, Y)
rm(LCS_cache, envir = .GlobalEnv)
return(list(X = X, Y = Y, lcs = sequence, length = l(sequence)))
}
Every call to LCS_recursive is made through the do.call function, which takes a list of arguments \( X \) and a function name \( f \), and returns \( f(X) \). do.call is useful when you want to supply arguments as a list as opposed to the standard syntax.
LCS_recursive <- function(X, Y, i = length(X), j = length(Y)) {
if (i == 0 || j == 0)
return()
if (X[i] == Y[j]) {
arguments <- list(X = X, Y = Y, i = i - 1, j = j - 1)
return(c(do.call("memoise", list(fun = "LCS_recursive", arguments = arguments),
), X[i]))
} else {
arguments_1 <- list(X = X, Y = Y, i = i, j = j - 1)
arguments_2 <- list(X = X, Y = Y, i = i - 1, j = j)
seq_length <- c(l(do.call("memoise", list(fun = "LCS_recursive", arguments = arguments_1))),
l(do.call("memoise", list(fun = "LCS_recursive", arguments = arguments_2))))
}
if (which.max(seq_length) == 1) {
return(do.call("memoise", list(fun = "LCS_recursive", arguments = arguments_1)))
} else {
return(do.call("memoise", list(fun = "LCS_recursive", arguments = arguments_2)))
}
}
The memoise is very simple; if LCS_recursive is called with i and j values identical to those used before, the result is returned from an array instead of by lengthy computation.
memoise <- function(fun, arguments) {
if (!exists("LCS_cache")) {
assign("LCS_cache", matrix(NA, l(arguments$X) + 2, l(arguments$Y) +
2), envir = .GlobalEnv)
}
i_ind <- arguments$i + 1
j_ind <- arguments$j + 1
if (!is.na(LCS_cache[i_ind, j_ind])) {
return(as.numeric(unlist(strsplit(LCS_cache[i_ind, j_ind], ","))))
} else {
LCS_cache[i_ind, j_ind] <<- paste(do.call("LCS_recursive", arguments),
collapse = ",")
return(as.numeric(unlist(strsplit(LCS_cache[i_ind, j_ind], ","))))
}
}
I'm going to use the (convieniently short) insulin protein sequence of ten different animals, and try to estimate the evolutionary similarity of the sequences to each other. This is a bit dubious; there are better measure of biological similarity than the longest common subsequence[1]
The sequences are from NCBI Protein; each letter denotes a particular amino acid. For the LCS function to work I need to swap these letters for numbers between one and twenty[2].
insulin_data <- list(guinea_pig = "MALWMHLLTVLALLALWGPNTNQAFVSRHLCGSNLVETLYSVCQDDGFFYIPKDRRELEDPQVEQTELGMGLGAGGLQPLALEMALQKRGIVDQCCTGTCTRHQLQSYCN",
brush_tailed_rat = "MAPWMHLLTVLALLALWGPNSVQAYSSQHLCGSNLVEALYMTCGRSGFYRPHDRRELEDLQVEQAELGLEAGGLQPSALEMILQKRGIVDQCCNNICTFNQLQNYCNVP",
zebrafish = "MAVWLQAGALLVLLVVSSVSTNPGTPQHLCGSHLVDALYLVCGPTGFFYNPKRDVEPLLGFLPPKSAQETEVADFAFKDHAELIRKRGIVEQCCHKPCSIFELQNYCN",
cod = "MAPPQHLCGSHLVDALYLVCGDRGFFYNPKGIVDQCCHRPCDIFDLQNYCN", hampster = "FVNQHLCGSHLVEALYLVCGERGFFYTPKSGIVDQCCTSICSLYQLENYCN",
sheep = "MALWTRLVPLLALLALWAPAPAHAFVNQHLCGSHLVEALYLVCGERGFFYTPKARREVEGPQVGALELAGGPGAGGLEGPPQKRGIVEQCCAGVCSLYQLENYCN",
rabbit = "MASLAALLPLLALLVLCRLDPAQAFVNQHLCGSHLVEALYLVCGERGFFYTPKSRREVEELQVGQAELGGGPGAGGLQPSALELALQKRGIVEQCCTSICSLYQLENYCN",
squirrel_monkey = "FVNQHLCGPHLVEALYLVCGERGFFYAPKTGVVDQCCTSICSLYQLQNYCN",
ostrich = "AANQHLCGSHLVEALYLVCGERGFFYSPKAGIVEQCCHNTCSLYQLENYCN", elephant = "FVNQHLCGSHLVEALYLVCGERGFFYTPKTGIVEQCCTGVCSLYQLENYCN")
amino_code <- list(G = 1, A = 2, V = 3, L = 4, I = 5, M = 6, F = 7,
W = 8, P = 9, S = 10, T = 11, C = 12, Y = 13, N = 14, Q = 15, D = 16, E = 17,
K = 18, R = 19, H = 20)
amino_acid_sequences <- lapply(X = insulin_data, function(x) {
count <- 1
temp <- unname(unlist(strsplit(x, "")))
for (i in names(amino_code)) {
temp[which(temp == i)] <- count
count <- count + 1
}
return(as.numeric(temp))
})
Next, I compute \( \frac{LCS}{length(X)+length(y)} \) as the metric of similarity, since the sequences vary in length relative to each other.
distance <- matrix(0, 10, 10)
for (i in 1:10) {
for (j in 1:i) {
X <- unlist(amino_acid_sequences[i])
Y <- unlist(amino_acid_sequences[j])
distance[i, j] <- LCS(X, Y)$length/(sum(length(X), length(Y)))
}
}
diag(distance) <- 0
rownames(distance) <- colnames(distance) <- names(insulin_data)
Finally, I plot a circular[3] phylogenetic tree of the distance data using the ape package.
require(ape)
scale = c("#A6CEE3", "#A6CEE3", "#1F78B4", "#1F78B4", "#A6CEE3", "#A6CEE3", "#A6CEE3", "#A6CEE3", "#B2DF8A", "#A6CEE3")
plot(as.phylo(hclust(dist(distance))), type = "fan", font = 2, cex = 0.8, sub = "lol", tip.col = scale)
title("phylo. tree based on insulin protein dissimilarity")
mtext("longest common subsequence")
[1] especially given than some amino acid substitutions have a larger effect on the protein function than others
[2] insulin is made up of standard proteinogenic amino-acids; no selenocysteine to confuse things here
[3] there are good reasons to only use circular phylogenetic trees