R BioConductor - Some Basics
Rowen Remis R. Iral
December 14, 2015
setwd("E:\\Other Business\\R User Meetup Philippines\\2015-12-16")
getwd()## [1] "E:/Other Business/R User Meetup Philippines/2015-12-16"R BioConductor - Basics
Rowen Remis R. Iral http://wenup.wordpress.com
R Users Group Philippines
BioConductor is a package that is designed for bioinformatics / genetics.
- Similar to devtools package
- Have its own package resources like CRAN
BioConductor Caveats
Bioconductor depends its installation version based on what R version you have.
BioInformatics
- the science of collecting and analyzing complex biological data such as genetic codes.
Genetics
- the study of heredity and the variation of inherited characteristics.
- the genetic properties or features of an organism, characteristic, etc
Genomics
- the branch of molecular biology concerned with the structure, function, evolution, and mapping of genomes.
Genomes
- the complete set of genes or genetic material present in a cell or organism.
Cells
- -
- -
- -
- -
- -
-
Images
Links: -http://people.eku.edu/ritchisong/cell1.gif -http://ocw.mit.edu/courses/biology/7-60-cell-biology-structure-and-functions-of-the-nucleus-spring-2010/7-60s10.jpg -http://mappingignorance.org/fx/media/2013/03/Fig1.png -https://upload.wikimedia.org/wikipedia/commons/thumb/e/e4/DNA_chemical_structure.svg/300px-DNA_chemical_structure.svg.png -http://i.livescience.com/images/i/000/053/587/original/dna-rna-structure.jpg?1370549225 -http://www.shubroto.com/sds-blogger/wp-content/uploads/2015/02/0323_DNA_Replication.jpg
{kind=link}
{kind=link}
{kind=link}
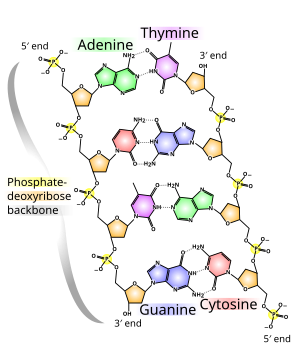{kind=link}
{kind=link}
{kind=link}
Other Utilities and Tools for BioInformatics / Genomics
- Tuxedo Suite (tophat, bowtie, cuffmerge, cufflinks, cummeRbund) - for Microarray Data
- Galaxy - for Workflow biomedical analysis
- Linux utilities like grep, wc, ls and others
- BAM, SAM and other tools, bamtools, samtools
- Python, BioPython
- R, Bioconductor
- Perl, Bioperl
- Integrated Genome Browser, Integrated Genome Viewer (IGB, IGV)
Manufacturers for DNA Sequencing Machines
- Roche
- Illumina (most popular)
- Life Technologies
- Beckman Coulter
- Pacific Biosciences
Using R Bioconductor
Packages to Install
source("http://bioconductor.org/biocLite.R")
biocLite()
biocLite(c("GenomeGraphs","beadarray","genefilter","Biostrings","BSgenome","biomaRt"))
biocLite(c("Biostrings","BSgenome","ShortRead","hwriter","seqinr","seqLogo"))
install.packages("ggplot2")
install.packages("RCurl")
install.packages("XML")
biocLite(c("GenomicFeatures","IRanges", "GenomicRanges", "ggbio", "biovizBase"))
library(BiocInstaller)
biocLite("XML")
biocLite("quantsmooth")Load Libraries
library(Biostrings)## Loading required package: BiocGenerics
## Loading required package: parallel
##
## Attaching package: 'BiocGenerics'
##
## The following objects are masked from 'package:parallel':
##
## clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
## clusterExport, clusterMap, parApply, parCapply, parLapply,
## parLapplyLB, parRapply, parSapply, parSapplyLB
##
## The following objects are masked from 'package:stats':
##
## IQR, mad, xtabs
##
## The following objects are masked from 'package:base':
##
## anyDuplicated, append, as.data.frame, as.vector, cbind,
## colnames, do.call, duplicated, eval, evalq, Filter, Find, get,
## grep, grepl, intersect, is.unsorted, lapply, lengths, Map,
## mapply, match, mget, order, paste, pmax, pmax.int, pmin,
## pmin.int, Position, rank, rbind, Reduce, rownames, sapply,
## setdiff, sort, table, tapply, union, unique, unlist, unsplit
##
## Loading required package: S4Vectors
## Loading required package: stats4
## Loading required package: IRanges
## Loading required package: XVectorBiostring Characters
# some commands
IUPAC_CODE_MAP## A C G T M R W S Y K
## "A" "C" "G" "T" "AC" "AG" "AT" "CG" "CT" "GT"
## V H D B N
## "ACG" "ACT" "AGT" "CGT" "ACGT"DNA_ALPHABET## [1] "A" "C" "G" "T" "M" "R" "W" "S" "Y" "K" "V" "H" "D" "B" "N" "-" "+"
## [18] "."DNA_BASES## [1] "A" "C" "G" "T"RNA_ALPHABET## [1] "A" "C" "G" "U" "M" "R" "W" "S" "Y" "K" "V" "H" "D" "B" "N" "-" "+"
## [18] "."RNA_BASES## [1] "A" "C" "G" "U"DNA Strings and RNA Strings
a = DNAString('ATGCCT')
a## 6-letter "DNAString" instance
## seq: ATGCCTclass(a)## [1] "DNAString"
## attr(,"package")
## [1] "Biostrings"b=RNAString('AUGCUGC')
b## 7-letter "RNAString" instance
## seq: AUGCUGCclass(b)## [1] "RNAString"
## attr(,"package")
## [1] "Biostrings"Alphabet Frequency
alphabetFrequency(a)## A C G T M R W S Y K V H D B N - + .
## 1 2 1 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0alphabetFrequency(a, baseOnly=TRUE)## A C G T other
## 1 2 1 2 0letterFrequency(a,"C")## C
## 2letterFrequency(a,"CG")## C|G
## 3Complementing Sequence
a## 6-letter "DNAString" instance
## seq: ATGCCTcomplement(a)## 6-letter "DNAString" instance
## seq: TACGGASearching sequence characters
a=DNAString("ACGTACGTACGC")
matchPattern("CGT", a, max.mismatch=1)## Views on a 12-letter DNAString subject
## subject: ACGTACGTACGC
## views:
## start end width
## [1] 2 4 3 [CGT]
## [2] 6 8 3 [CGT]
## [3] 10 12 3 [CGC]countPattern("CGT", a, max.mismatch=1)## [1] 3Genome Resources
library(BSgenome)## Loading required package: GenomeInfoDb
## Loading required package: GenomicRanges
## Loading required package: rtracklayeravailable.genomes()## [1] "BSgenome.Alyrata.JGI.v1"
## [2] "BSgenome.Amellifera.BeeBase.assembly4"
## [3] "BSgenome.Amellifera.UCSC.apiMel2"
## [4] "BSgenome.Amellifera.UCSC.apiMel2.masked"
## [5] "BSgenome.Athaliana.TAIR.04232008"
## [6] "BSgenome.Athaliana.TAIR.TAIR9"
## [7] "BSgenome.Btaurus.UCSC.bosTau3"
## [8] "BSgenome.Btaurus.UCSC.bosTau3.masked"
## [9] "BSgenome.Btaurus.UCSC.bosTau4"
## [10] "BSgenome.Btaurus.UCSC.bosTau4.masked"
## [11] "BSgenome.Btaurus.UCSC.bosTau6"
## [12] "BSgenome.Btaurus.UCSC.bosTau6.masked"
## [13] "BSgenome.Btaurus.UCSC.bosTau8"
## [14] "BSgenome.Celegans.UCSC.ce10"
## [15] "BSgenome.Celegans.UCSC.ce2"
## [16] "BSgenome.Celegans.UCSC.ce6"
## [17] "BSgenome.Cfamiliaris.UCSC.canFam2"
## [18] "BSgenome.Cfamiliaris.UCSC.canFam2.masked"
## [19] "BSgenome.Cfamiliaris.UCSC.canFam3"
## [20] "BSgenome.Cfamiliaris.UCSC.canFam3.masked"
## [21] "BSgenome.Dmelanogaster.UCSC.dm2"
## [22] "BSgenome.Dmelanogaster.UCSC.dm2.masked"
## [23] "BSgenome.Dmelanogaster.UCSC.dm3"
## [24] "BSgenome.Dmelanogaster.UCSC.dm3.masked"
## [25] "BSgenome.Dmelanogaster.UCSC.dm6"
## [26] "BSgenome.Drerio.UCSC.danRer5"
## [27] "BSgenome.Drerio.UCSC.danRer5.masked"
## [28] "BSgenome.Drerio.UCSC.danRer6"
## [29] "BSgenome.Drerio.UCSC.danRer6.masked"
## [30] "BSgenome.Drerio.UCSC.danRer7"
## [31] "BSgenome.Drerio.UCSC.danRer7.masked"
## [32] "BSgenome.Ecoli.NCBI.20080805"
## [33] "BSgenome.Gaculeatus.UCSC.gasAcu1"
## [34] "BSgenome.Gaculeatus.UCSC.gasAcu1.masked"
## [35] "BSgenome.Ggallus.UCSC.galGal3"
## [36] "BSgenome.Ggallus.UCSC.galGal3.masked"
## [37] "BSgenome.Ggallus.UCSC.galGal4"
## [38] "BSgenome.Ggallus.UCSC.galGal4.masked"
## [39] "BSgenome.Hsapiens.1000genomes.hs37d5"
## [40] "BSgenome.Hsapiens.NCBI.GRCh38"
## [41] "BSgenome.Hsapiens.UCSC.hg17"
## [42] "BSgenome.Hsapiens.UCSC.hg17.masked"
## [43] "BSgenome.Hsapiens.UCSC.hg18"
## [44] "BSgenome.Hsapiens.UCSC.hg18.masked"
## [45] "BSgenome.Hsapiens.UCSC.hg19"
## [46] "BSgenome.Hsapiens.UCSC.hg19.masked"
## [47] "BSgenome.Hsapiens.UCSC.hg38"
## [48] "BSgenome.Hsapiens.UCSC.hg38.masked"
## [49] "BSgenome.Mfascicularis.NCBI.5.0"
## [50] "BSgenome.Mfuro.UCSC.musFur1"
## [51] "BSgenome.Mmulatta.UCSC.rheMac2"
## [52] "BSgenome.Mmulatta.UCSC.rheMac2.masked"
## [53] "BSgenome.Mmulatta.UCSC.rheMac3"
## [54] "BSgenome.Mmulatta.UCSC.rheMac3.masked"
## [55] "BSgenome.Mmusculus.UCSC.mm10"
## [56] "BSgenome.Mmusculus.UCSC.mm10.masked"
## [57] "BSgenome.Mmusculus.UCSC.mm8"
## [58] "BSgenome.Mmusculus.UCSC.mm8.masked"
## [59] "BSgenome.Mmusculus.UCSC.mm9"
## [60] "BSgenome.Mmusculus.UCSC.mm9.masked"
## [61] "BSgenome.Osativa.MSU.MSU7"
## [62] "BSgenome.Ptroglodytes.UCSC.panTro2"
## [63] "BSgenome.Ptroglodytes.UCSC.panTro2.masked"
## [64] "BSgenome.Ptroglodytes.UCSC.panTro3"
## [65] "BSgenome.Ptroglodytes.UCSC.panTro3.masked"
## [66] "BSgenome.Rnorvegicus.UCSC.rn4"
## [67] "BSgenome.Rnorvegicus.UCSC.rn4.masked"
## [68] "BSgenome.Rnorvegicus.UCSC.rn5"
## [69] "BSgenome.Rnorvegicus.UCSC.rn5.masked"
## [70] "BSgenome.Rnorvegicus.UCSC.rn6"
## [71] "BSgenome.Scerevisiae.UCSC.sacCer1"
## [72] "BSgenome.Scerevisiae.UCSC.sacCer2"
## [73] "BSgenome.Scerevisiae.UCSC.sacCer3"
## [74] "BSgenome.Sscrofa.UCSC.susScr3"
## [75] "BSgenome.Sscrofa.UCSC.susScr3.masked"
## [76] "BSgenome.Tgondii.ToxoDB.7.0"
## [77] "BSgenome.Tguttata.UCSC.taeGut1"
## [78] "BSgenome.Tguttata.UCSC.taeGut1.masked"
## [79] "BSgenome.Tguttata.UCSC.taeGut2"
## [80] "BSgenome.Vvinifera.URGI.IGGP12Xv0"
## [81] "BSgenome.Vvinifera.URGI.IGGP12Xv2"available.SNPs()## [1] "SNPlocs.Hsapiens.dbSNP.20090506"
## [2] "SNPlocs.Hsapiens.dbSNP.20100427"
## [3] "SNPlocs.Hsapiens.dbSNP.20101109"
## [4] "SNPlocs.Hsapiens.dbSNP.20110815"
## [5] "SNPlocs.Hsapiens.dbSNP.20111119"
## [6] "SNPlocs.Hsapiens.dbSNP.20120608"
## [7] "SNPlocs.Hsapiens.dbSNP141.GRCh38"
## [8] "SNPlocs.Hsapiens.dbSNP142.GRCh37"
## [9] "SNPlocs.Hsapiens.dbSNP144.GRCh37"
## [10] "SNPlocs.Hsapiens.dbSNP144.GRCh38"
## [11] "XtraSNPlocs.Hsapiens.dbSNP141.GRCh38"
## [12] "XtraSNPlocs.Hsapiens.dbSNP144.GRCh37"
## [13] "XtraSNPlocs.Hsapiens.dbSNP144.GRCh38"Read the Dengue FASTA File
library(ShortRead)## Loading required package: BiocParallel
## Loading required package: Rsamtools
## Loading required package: GenomicAlignments
## Loading required package: SummarizedExperiment
## Loading required package: Biobase
## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.dengue.fa <- readFasta(".", "1K4R.fasta")## Warning in .Call2("fasta_index", filexp_list, nrec, skip, seek.first.rec, :
## reading FASTA file ./1K4R.fasta.txt: ignored 315 invalid one-letter
## sequence codesdengue.fa## class: ShortRead
## length: 3 reads; width: 290 cyclessread(dengue.fa)## A DNAStringSet instance of length 3
## width seq
## [1] 290 SRCTHNRDVTGTGTTRVTVGGCVTTAGKSMD...AVAHGSDVNVAMTNTNNGGGMGDNYVGSHWK
## [2] 290 SRCTHNRDVTGTGTTRVTVGGCVTTAGKSMD...AVAHGSDVNVAMTNTNNGGGMGDNYVGSHWK
## [3] 290 SRCTHNRDVTGTGTTRVTVGGCVTTAGKSMD...AVAHGSDVNVAMTNTNNGGGMGDNYVGSHWKid(dengue.fa)## A BStringSet instance of length 3
## width seq
## [1] 27 1K4R:A|PDBID|CHAIN|SEQUENCE
## [2] 27 1K4R:B|PDBID|CHAIN|SEQUENCE
## [3] 27 1K4R:C|PDBID|CHAIN|SEQUENCEGet Frequencies of each sequence
alphabetFrequency(sread(dengue.fa)[[1]])## A C G T M R W S Y K V H D B N - + .
## 32 12 36 37 9 14 8 16 10 28 36 17 20 0 15 0 0 0alphabetFrequency(sread(dengue.fa)[[2]])## A C G T M R W S Y K V H D B N - + .
## 32 12 36 37 9 14 8 16 10 28 36 17 20 0 15 0 0 0alphabetFrequency(sread(dengue.fa)[[3]])## A C G T M R W S Y K V H D B N - + .
## 32 12 36 37 9 14 8 16 10 28 36 17 20 0 15 0 0 0alphabetFrequency(sread(dengue.fa)[[1]], baseOnly=TRUE)## A C G T other
## 32 12 36 37 173alphabetFrequency(sread(dengue.fa)[[2]], baseOnly=TRUE)## A C G T other
## 32 12 36 37 173alphabetFrequency(sread(dengue.fa)[[3]], baseOnly=TRUE)## A C G T other
## 32 12 36 37 173Getting Data from NCBI
Get NCBI Sequence custom function
getncbiseq <- function(accession) {
require("seqinr") # this function requires the SeqinR R package
# first find which ACNUC database the accession is stored in:
dbs <- c("genbank","refseq","refseqViruses","bacterial")
numdbs <- length(dbs)
for (i in 1:numdbs) {
db <- dbs[i]
choosebank(db)
# check if the sequence is in ACNUC database â???Tdbâ???T:
resquery <- try(query(".tmpquery", paste("AC=", accession)), silent = TRUE)
if (!(inherits(resquery, "try-error")))
{
print(paste("trying: ",db))
queryname <- "query2"
thequery <- paste("AC=",accession,sep="")
print(thequery)
# query("queryname","thequery")
query2 <- query(`queryname`,`thequery`)
# see if a sequence was retrieved:
seq <- getSequence(query2$req[[1]])
closebank()
return(seq)
}
print(paste("accession not in: ",db))
closebank()
}
print(paste("ERROR: accession",accession,"was not found"))
}Get the Dengue Virus 1 Sequence (1 of 4 variants)
dengueseq <- getncbiseq("NC_001477")## Loading required package: seqinr
## Loading required package: ade4
##
## Attaching package: 'ade4'
##
## The following object is masked from 'package:BSgenome':
##
## score
##
## The following object is masked from 'package:rtracklayer':
##
## score
##
## The following object is masked from 'package:GenomicRanges':
##
## score
##
## The following object is masked from 'package:Biostrings':
##
## score
##
## The following object is masked from 'package:IRanges':
##
## score
##
## The following object is masked from 'package:BiocGenerics':
##
## score
##
##
## Attaching package: 'seqinr'
##
## The following object is masked from 'package:Biostrings':
##
## translate## [1] "accession not in: genbank"
## [1] "accession not in: refseq"
## [1] "trying: refseqViruses"
## [1] "AC=NC_001477"Write to Fasta File den1.fasta
write.fasta(names="DEN-1", sequences=dengueseq, file.out="den1.fasta")Reading data back from FASTA file
library("seqinr")
dengue <- read.fasta(file = "den1.fasta")
# Get the Vector of Nucleotide sequence
dengueseq <- dengue[[1]]Get Length of Dengue Sequence
length(dengueseq)## [1] 10735Count Bases
table(dengueseq)## dengueseq
## a c g t
## 3426 2240 2770 2299GC-content (Guanine-Cytosine Content)
GC-Content Percentage - In molecular biology and genetics, GC-content (or guanine-cytosine content) is the percentage of nitrogenous bases on a DNA molecule that are either guanine or cytosine (from a possibility of four different ones, also including adenine and thymine).
\(\frac{G + C}{A + T + G + C}\)
And the Ratio is:
\(\frac{A + T}{G + C}\)
GC Function
GC(dengueseq)## [1] 0.4666977The result above means that the fraction of bases in the DEN-1 Dengue virus genome that are Gs or Cs. Multiply to 100 to get the percentage.
checking
tab <- table(dengueseq)
(tab["g"]+tab["c"])/sum(tab)## g
## 0.4666977Get DNA words Counts that are 1 nucleotide long
count(dengueseq, 1)##
## a c g t
## 3426 2240 2770 2299On 2 nucleotide long
count(dengueseq, 2)##
## aa ac ag at ca cc cg ct ga gc gg gt ta tc tg
## 1108 720 890 708 901 523 261 555 976 500 787 507 440 497 832
## tt
## 529Other Reference and Resources:
- http://web1.sph.emory.edu/users/hwu30/teaching/bioc/biostrings.pdf
- http://cs.mcgill.ca/~blanchem/561/GenBio.pdf
- http://groups.csail.mit.edu/medg/courses/6872/2003/slides/mit.pdf
- http://www.nature.com/scitable/ebooks/genomics-4/
- http://www.yourgenome.org/facts/what-is-dna
- https://github.com/avrilcoghlan/LittleBookofRBioinformatics
- http://a-little-book-of-r-for-bioinformatics.readthedocs.org/en/latest/src/chapter1.html
- http://www.cs.ukzn.ac.za/~hughm/bio/scripts/script_2_accessing_sequences.R
- https://blackberryaurora.wordpress.com/2015/01/21/simple-r-functions/
- http://www.rcsb.org/pdb/explore.do?structureId=1k4r
- https://en.wikipedia.org/wiki/GC-content
- https://en.wikipedia.org/wiki/DNA_sequencer
SessionInfo
sessionInfo()## R version 3.2.3 (2015-12-10)
## Platform: x86_64-w64-mingw32/x64 (64-bit)
## Running under: Windows 7 x64 (build 7601) Service Pack 1
##
## locale:
## [1] LC_COLLATE=English_United States.1252
## [2] LC_CTYPE=English_United States.1252
## [3] LC_MONETARY=English_United States.1252
## [4] LC_NUMERIC=C
## [5] LC_TIME=English_United States.1252
##
## attached base packages:
## [1] stats4 parallel stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] seqinr_3.1-3 ade4_1.7-3
## [3] ShortRead_1.28.0 GenomicAlignments_1.6.1
## [5] SummarizedExperiment_1.0.1 Biobase_2.30.0
## [7] Rsamtools_1.22.0 BiocParallel_1.4.0
## [9] BSgenome_1.38.0 rtracklayer_1.30.1
## [11] GenomicRanges_1.22.2 GenomeInfoDb_1.6.1
## [13] Biostrings_2.38.2 XVector_0.10.0
## [15] IRanges_2.4.6 S4Vectors_0.8.5
## [17] BiocGenerics_0.16.1
##
## loaded via a namespace (and not attached):
## [1] knitr_1.11 magrittr_1.5 zlibbioc_1.16.0
## [4] lattice_0.20-33 hwriter_1.3.2 stringr_1.0.0
## [7] tools_3.2.3 grid_3.2.3 latticeExtra_0.6-26
## [10] lambda.r_1.1.7 futile.logger_1.4.1 htmltools_0.2.6
## [13] yaml_2.1.13 digest_0.6.8 RColorBrewer_1.1-2
## [16] futile.options_1.0.0 bitops_1.0-6 RCurl_1.95-4.7
## [19] evaluate_0.8 rmarkdown_0.8.1 stringi_1.0-1
## [22] BiocInstaller_1.20.1 XML_3.98-1.3