バイオインフォマティクス解析におけるRの実践的基礎
Yusuke Matsui
Biomedical and Health Informatics Unit (BMHI),
Department of Integrated Health Sciences,
Graduate School of Medicine,
Nagoya University, Systems Biology Division,
Integrated Glycan-Big Data Center (iGDATA),
Institute for Glycobiology Core Research
(iGCORE)
2024-10-09
なぜRをあえて「講義」で学ぶのか?
バイオインフォマティクス解析はRのみで完結することはない。シェルコマンド(+awk、sed等)やPythonなどを適材適所で組み合わせ、全体で一つの解析フローを構築することが一般的である。
R vs Python: 二元論に意味はあるのか?
近年、深層学習モデルの構築において柔軟に対応できるPythonがRより優位であるという意見が頻繁に見受けられる。企業に就職した学生からも「RをやめてPythonを使うようになった」と、Pythonが優れているかのような話を聞くことがある。しかし、実際にはどちらのコミュニティも活発に開発が続けられており、膨大なコード、パッケージ、概念が蓄積されている。RとPythonそれぞれに強みと弱みがあることを考えれば、二元論的な見方に意味はないと言えるだろう(Rユーザーの強がりかもしれないが)。
AI時代のR学習: その意義とは?
インターネット上には、Rに関する無数の体系的・断片的な知識が存在し、ChatGPTなどのAIツールを使えばやりたいことを即座にコードに変換してくれる時代である。そのような時代に、あえてRを講義形式で学ぶ意義はあるのだろうか?(話している私自身も)その疑問を抱くことがある。知らないことがあれば、ネットで調べるか、AIツールに聞けば解決することが多いのは確かだ。
Rを学ぶ上で「講義形式」が重要な理由
それでも、何か足りないものがあるとすれば、それは何か?という問いを(教える側としては)考えざるを得ない。特に初心者にとって、ゼロからイチへ進む壁は大きい。その要因の一つとして、全体像が見えにくいことが挙げられるだろう。しかし、全体像を説明されても具体的なイメージがわかず、個別のコードを学んでも使いどころがわからないという状況に陥ることがある。そうしているうちに、他の選択肢に目が向き、「本当にRでいいのか?」(Pythonへの誘惑など)と考え始め、モチベーションを保てなくなることも多い。一方、ネット上のブログや記事は、特定の目的を解決するためのコード例が主体であり、それをコピペして使うだけという学習スタイルが多く見受けられる。
講義形式で学ぶことの意義: 体系的な知識と実践的スキル
どのように学ぶのが適切なのかについて、確固たる正解は存在しない。しかし、少なくともRを講義形式で学ぶことの意義は、「体系的な知識の習得」と「実践的なスキルの習得」にあると考える。インターネットには、単発的な解決策や断片的な情報が溢れているが、それらを繋ぎ合わせて「どのように一貫した解析を行うか」という視点が欠けていることが多い。データの取り扱い、前処理、探索的データ解析(EDA)、統計解析や機械学習、結果の解釈と可視化まで、一連の流れを通じて見通しを持つことは極めて重要である。
実際の解析シナリオでのRの活用法を学ぶ
例えば、Rでの解析は、多くのパッケージを活用することで自動化や効率化を図ることが可能である。tidyverse
パッケージ群を用いたデータ操作、ggplot2
を用いた高品質な可視化、dplyr や data.table
を用いたデータフレームの操作などは、単一の操作に留まらず、他の関数と組み合わせて強力なデータ解析パイプラインを構築できる。しかし、これらの関数やパッケージの使い方を単独で学んでも、その役割や全体での連携が理解できなければ、知識が断片化し、効果的に活用できなくなる。
講義の目的:Rを通じてデータ解析の全体像を把握する
この講義が目指すのは、「データ解析の全体像を示し、その中でRを使った各ステップがどのように役立つかを具体的に理解すること」である。バイオインフォマティクス解析の中でRが持つ重要な役割と、他ツールとの連携を意識しながら、実践的な解析フローの中でRを最大限に活用できる力を身につけることが目的だ。
Rがバイオインフォマティクスで活用される場面
Rの役割と他ツールとの連携の重要性
バイオインフォマティクス解析では、Rはその強力な統計解析機能や可視化能力によって広範囲に活用されるが、Rだけで全体の解析が完結することは稀である。シェルコマンドやPythonなどのツールと連携しながら、解析フローを構築するのが一般的だ。ここでは、RNA-seq解析を例に取り、Rがどのように役立ち、他のツールとどのように連携するかを見ていく。
例:RNA-seq解析におけるRの具体的な役割
RNA-seq解析は、データの前処理から発現変動解析、結果の可視化まで、多段階にわたるフローが必要だ。以下では、その各ステップでRの果たす役割と、それを補完する他のツールについて解説する。以下は、nf-core/rnaseqパイプラインにおける各処理ステップ、使用ツール、処理の目的、対応するスクリプトやプログラム言語を表形式にまとめたものである。
| 処理ステップ | 使用ツール | 目的 | スクリプト/プログラム言語 |
|---|---|---|---|
| FastQファイルの結合 | cat |
リシーケンスされたFastQファイルをサンプルごとに結合 | シェルスクリプト |
| FastQファイルのサブサンプリングとストランド推定 | fq, Salmon | サブサンプリングしてライブラリのストランド情報を推定 | Python, シェル |
| リードの品質管理 (QC) | FastQC | シーケンスリードの品質を評価 | シェルスクリプト |
| UMI抽出 | UMI-tools | UMIを抽出してPCR重複を補正 | シェルスクリプト |
| アダプター除去と品質トリミング | Trim Galore! | アダプター除去と低品質リードのトリミング | シェルスクリプト |
| ゲノムコンタミナントの除去 | BBSplit | ゲノムに由来するコンタミナントの除去 | シェルスクリプト |
| リボソームRNAの除去 | SortMeRNA | リボソームRNAの除去 | シェルスクリプト |
| アライメントと定量化ルートの選択 | STAR, Salmon, RSEM, HiSAT2 | リードのアライメントと定量化 | シェルスクリプト |
| アライメントのソートとインデックス作成 | SAMtools | アライメントファイルのソートとインデックス作成 | シェルスクリプト |
| UMIベースの重複除去 | UMI-tools | UMIに基づいてPCRの重複リードを除去 | シェルスクリプト |
| トランスクリプト組み立てと定量化 | StringTie | トランスクリプトアセンブリと発現量の定量化 | シェルスクリプト |
| 広範な品質管理 (QC) | RSeQC, Qualimap, dupRadar, Preseq, DESeq2 | 発現データの品質評価と異常やバイアスの検出 | R, シェルスクリプト |
| 発現変動解析 | DESeq2 | 発現変動遺伝子の解析、統計モデルの構築 | R | | |||
| 結果の可視化とQCのレポート | MultiQC, R | QC結果、発現量の可視化、サンプル間類似度の評価 | R, シェルスクリプト |
ここでは、それぞれが以下の役割を担っている。シェルスクリプトが多数を占めているように見えるが、それぞれのソフトウェアを呼び出すのに効率的であるためである。
シェルスクリプト: 大量データの処理、ファイル操作、並列処理に使用され、ファイルの結合、トリミング、除去などの高速操作を担当。
Python: データの前処理、サブサンプリング、サンプルの準備段階での操作に適用。主に柔軟なデータ操作と効率的なスクリプト実行が必要な場面で使用。
R: 最終的な解析と可視化において使用。特に発現変動解析(DESeq2)やQCの結果をまとめて可視化する場面で重要な役割を果たす。
このようなパイプラインにおけるRの役割は、データ解析と可視化の最終ステップに位置づけられることがわかる。特に次のような場面で重要である:
- 発現変動解析:
- ツール: DESeq2
- Rは、発現変動解析の結果を導き出すために、モデル構築や統計的テストを実施する。DESeq2はRNA-seqデータ解析における代表的なツールで、R環境内で動作し、統計モデルを通じて遺伝子発現の変動を評価する。
- 結果の可視化:
- ツール: MultiQC, R
- 各ステップのQC結果や発現量の分布、サンプル間の類似度など、Rを使用してデータの視覚化が行われる。Rのグラフィックライブラリ(ggplot2など)を使用して、データを直感的に把握するための図が作成される。
Rがバイオインフォマティクスで活躍できる場面と限界のある場面
上記のRNA-seq解析のフローはバイオインフォマティクス解析の一例であり、Rが活躍する場面が限られているように見えるかもしれない。もちろん、シェルやPythonで行う多くのタスクはRでも可能だが、以下のようにRのシステム的な制約や効率性を考慮すると、必ずしもRが最適な選択肢とは言えない場合がある。
大規模データ処理におけるRの限界
Rは大規模データの取り扱いにおいて、メモリ使用量や実行速度に制約がある。特に、数百ギガバイト単位のデータを読み込み、前処理する場合には、シェルやPythonが優れたパフォーマンスを発揮する。data.tableやdplyrといったRパッケージを活用すれば、ある程度のパフォーマンス向上は可能であるが、膨大なデータを効率的に操作するには、シェルやPythonの方が適している。
Rの強みを最大限に活かす場面
一方で、Rの強力な統計解析機能とデータ可視化能力は他の言語にはない強みである。特に、ggplot2やshinyを用いたデータの高度な可視化や、インタラクティブなデータ分析において、Rは非常に有効である。これらの特性が必要な場面では、Rを用いることで他言語にない精度と表現力を発揮できる。
適材適所でツールを使い分ける
バイオインフォマティクス解析では、各ツールの特性を活かして適材適所で使い分けることが求められる。シェルはファイル操作や大規模データの分割・統合、Pythonは機械学習モデルの構築や柔軟なデータ操作、そしてRは高度な統計解析や視覚化に強みがある。このように、ツール間で役割を分担することで、解析の効率化と精度を両立させることができる。
解析フローの設計
解析フローを最適化するためには、ツール間のデータ形式の整合性を保ち、統一した解析パイプラインを構築することが重要である。例えば、Rでデータの前処理を行い、その結果をPythonでモデル化する、あるいは逆にPythonで前処理したデータをRで統計解析するといったアプローチも可能だ。このように、R、シェル、Pythonの特性を理解し、最も適したツールを柔軟に組み合わせることで、複雑な解析タスクに対応できる。
R、シェル、Pythonを組み合わせる際のベストプラクティス
- 適材適所でツールを選択:
- データ前処理やファイル操作にはシェル、高度なデータ操作や機械学習にはPython、統計解析や可視化にはRを用いる。
- 再現性の確保:
- 解析手順をスクリプト化し、
snakemakeやnextflowなどを活用して再現性を高める。解析結果をrmarkdownやjupyter notebookでドキュメント化し、共有可能な形でプロジェクト全体を管理する。
- 解析手順をスクリプト化し、
- 環境管理:
condaを使用して、依存関係を管理し、再現性のある解析環境を構築する。Dockerコンテナを用いて解析環境を統一し、他の研究者とも共有しやすくする。
Rの役割は下流解析において重要
生データを定量値に変換するプロセスでは、Rの役割は比較的小さい場合が多い。これは、データの前処理や品質管理などが多くの解析パイプラインで自動化され、複数のツールが連携して処理されるためである。しかし、定量アッセイ全般における下流解析では、Rが非常に重要な役割を果たす。この段階では、複数のデータの統合、アノテーション、メタ情報の処理などが含まれ、定量されたデータを知識へと変換するプロセスが進行する。
Rを用いてできることの全貌
ここで、Rがどれだけ幅広く活用できるか、その全貌を見ていくことにしよう。Rは、バイオインフォマティクス解析に限らず、以下のようにさまざまなデータ解析や統計モデリングに応用可能である。
| 項目 | 説明 | 具体例 |
|---|---|---|
| データの入出力と前処理 | CSV、Excel、JSON、SQLなどのデータを効率的に読み込む。欠損値処理やスケーリングも容易。 | read.csv, fread, tidyverse,
janitor |
| データの操作と変換 | データのフィルタリング、集計、結合やピボット操作が可能。文字列操作も効率的に行える。 | dplyr, data.table, tidyr,
stringr |
| データの可視化 | 基本的なプロットやカスタマイズ可能な高度な可視化が可能。インタラクティブなグラフ作成もサポート。 | plot, ggplot2, plotly,
shiny |
| 統計解析とモデリング | 基本的な統計解析や回帰モデル、ベイズ統計も扱える。混合効果モデルやサバイバル分析も可能。 | t.test, aov, glm,
lme4, rstan, brms |
| 機械学習とAI | 教師あり学習、教師なし学習、深層学習が可能。多くの機械学習アルゴリズムに対応。 | caret, randomForest, e1071,
keras, tensorflow |
| 時系列解析 | ARIMAモデルなどを使った時系列解析や異常検知が可能。 | forecast, ts |
| 空間データ解析 | 地理空間データの取り扱いや解析、地図の描画が可能。 | sf, sp, raster |
| テキストマイニングとNLP | テキストデータの前処理やトピックモデリング、感情分析、文章分類などを行える。 | tm, text2vec, LDA |
| リポート作成とプレゼン | rmarkdown
を使った自動レポート生成や、shiny
を用いたインタラクティブなダッシュボード作成が可能。 |
rmarkdown, shiny |
| プログラミングとアルゴリズム | 基本的なプログラミング操作に加え、Rパッケージの作成や関数の共有が可能。 | devtools, usethis |
Rを用いた下流解析
下流解析には、一般的に標準的な解析を実行するためのパッケージが存在する。バイオインフォマティクス解析では、主にBioconductorに登録されているパッケージが多くの役割を果たしている。これらのパッケージには、定量データを入力すると自動的に処理を行ってくれる高機能な関数も存在するが、多くの場合、解析フローをステップごとに組み立てる必要がある。各ステップで適切な関数を組み合わせて、全体の解析フローを構築するのが一般的だ。
Bioconductorパッケージを使うとできること
Bioconductorは、R環境でのバイオインフォマティクス解析を支援するための大規模なソフトウェアプロジェクトであり、特に次世代シーケンシング(NGS)やマイクロアレイデータなど、生命科学研究に特化した解析を行うためのパッケージを豊富に提供している。以下に、Bioconductorで行える主な解析内容を分野別にまとめた。
| 分析タイプ | 説明 | 主なパッケージ/ツール |
|---|---|---|
| 遺伝子発現解析 | マイクロアレイやRNA-seqデータの発現変動解析。パスウェイ解析や時系列解析も対応。 | affy, limma, DESeq2,
edgeR, tximport, DEXSeq,
clusterProfiler, ReactomePA |
| エピゲノム解析 | ChIP-seq、ATAC-seqデータのピークアノテーションや発現変動解析。
| ChIPseeker, DiffBind, csaw,
chromVAR, ATACseqQC |
|
| 遺伝子構造と変異解析 | ゲノムアノテーションとバリアント解析。構造変異や突然変異シグネチャ解析も対応。 | GenomicRanges, rtracklayer,
biomaRt, VariantAnnotation,
StructuralVariantAnnotation,
MutationalPatterns |
| メチル化解析 | DNAメチル化データやバイサルファイトシーケンスデータの解析。 | minfi, RnBeads, BiSeq,
methylKit |
| マイクロバイオーム解析 | メタゲノムや16S rRNAデータのインポート、正規化、可視化、解析。 | phyloseq, metagenomeSeq,
dada2, microbiome |
| シングルセル解析 | シングルセルRNA-seqデータの前処理、クラスタリング、発現変動解析。 | scater, scran,
SingleCellExperiment, Seurat,
Scanpy, monocle, MAST,
tradeSeq |
| 質量分析データ解析 | メタボロームやプロテオームデータのピーク検出、統合解析、タンデム質量スペクトル解析。 | XCMS, MetaboAnalystR, CAMERA,
MSnbase, ProteoMM, mzR,
proFIA, Spectra, mzIdentML |
| ネットワーク解析 | 遺伝子共発現ネットワークや相互作用ネットワークの解析と可視化。 | WGCNA, igraph, graph,
STRINGdb, biogrid, pathview |
| インタラクティブなデータ解析 | Webアプリケーションやシングルセルデータのインタラクティブな可視化と操作。 | shiny, iSEE |
| パイプラインとワークフロー | 再現性の高い解析ワークフローの構築とデータ管理。 | BiocWorkflowTools, workflowr,
Drake, ExperimentHub,
AnnotationHub, MultiAssayExperiment |
| 知識ベースとアノテーション | ゲノムや遺伝子のアノテーション、アノテーションデータの取得と管理。 | biomaRt, GenomicFeatures,
org.Hs.eg.db, AnnotationDbi,
EnsDb |
このように、Bioconductorを活用することで、バイオインフォマティクスのほぼ全ての解析プロセスを網羅的かつ効率的に行うことができる。各解析手法はRの基本的なデータ操作や統計解析の上に成り立っており、それらを組み合わせて高度な解析を行うことが可能である。これらの機能を知ることで、バイオインフォマティクス研究の現場でRとBioconductorパッケージ群をどのように活用するかの全体像が明確になるだろう。
Bioconductorの特筆すべき利点
- データの一貫した取り扱い:
- Bioconductorでは、
SummarizedExperimentやSingleCellExperimentなどの統一されたデータ構造を使用することで、解析の各ステップ間でのデータの受け渡しがスムーズになる。これにより、前処理から解析、可視化までを一貫したワークフローで効率的に進めることができる。
- Bioconductorでは、
- 豊富なアノテーション情報の統合:
- Bioconductorには、
biomaRtやAnnotationHubなどのパッケージがあり、遺伝子やタンパク質に関する豊富なアノテーション情報を簡単に取得・統合できる。これにより、解析結果を生物学的知識と結びつけて解釈するのが容易になる。
- Bioconductorには、
- 最新の解析手法への対応:
- Bioconductorは、RNA-seq、エピゲノム、シングルセルRNA-seqなど、最先端のオミクスデータに対応した解析ツールを提供している。例えば、
DESeq2やedgeRでの発現変動解析、scranやSeuratでのシングルセル解析、ChIPseekerでのChIP-seq解析など、最新の研究手法に迅速に対応している。
- Bioconductorは、RNA-seq、エピゲノム、シングルセルRNA-seqなど、最先端のオミクスデータに対応した解析ツールを提供している。例えば、
- コミュニティとサポート:
- Bioconductorは活発なコミュニティに支えられており、常に新しいパッケージや手法が開発・公開されている。公式サイトには詳細なドキュメントやチュートリアルが豊富に揃っており、解析手法を迅速に学ぶことができる。定期的なアップデートにより、常に最新の技術に触れることが可能だ。
- 再現性の高い解析:
- Bioconductorは再現性を重視しており、
BiocParallelを使った並列処理やBiocManagerによるパッケージ管理機能で、解析の再現性を確保している。これにより、他の研究者が同じ条件で解析を再現することが容易になる。
- Bioconductorは再現性を重視しており、
R + Bioconductorの強力な組み合わせ
RとBioconductorの組み合わせは、バイオインフォマティクス解析における非常に強力なツールセットを提供する。RNA-seq解析では、データのインポートから発現変動解析、アノテーション情報の付加、結果の可視化までを一貫して行うことができる。さらに、シングルセルRNA-seq解析やChIP-seq解析など、他のオミクスデータとの統合も容易で、解析フロー全体を効率化しつつ、より深い生物学的知見を得ることが可能である。
Bioconductorはまた、ggplot2やComplexHeatmapなどのRの可視化パッケージとシームレスに連携するため、解析結果を直感的に理解できる形式で表現できる。さらに、shinyを利用すればインタラクティブなアプリケーションを構築し、研究データをリアルタイムで探索・可視化することもできる。
このように、RとBioconductorを活用することで、バイオインフォマティクス解析は単なるデータ処理を超えて、仮説の検証や新しい発見を生むための非常に強力なツールとなる。Rは、その統計解析機能と可視化能力により、特に生物学的データの解析において大きな役割を果たす一方、シェルやPythonとの連携が解析フロー全体の効率化に欠かせない。
(補足)Pythonとの比較
Pythonには、RのBioconductorに相当するバイオインフォマティクス解析のための包括的なプラットフォームは確立していない。ただし、いくつかのライブラリやツールがその役割を担っている。これらのライブラリは、特定の解析タスクに対応するものであり、Bioconductorのように統合されたエコシステムと呼ぶには不十分かもしれない。たとえば、BiopythonはPythonにおけるバイオインフォマティクス解析のための強力なライブラリであり、シーケンスデータの操作や生物学的データの解析に広く利用されている。しかし、RとBioconductorを積極的に選ぶ理由には、次のような本質的な違いがある。
1. 統計解析とモデリングの強力なサポート
Rは、統計解析やモデリングにおいて非常に強力な基盤を持っている。バイオインフォマティクス解析における高度な統計的手法の適用に適しており、特にRNA-seq解析では、DESeq2
や edgeR
といったパッケージがその代表例である。これらのパッケージは、一般化線形モデル(GLM)やベイズ統計などの高度な手法を簡便に使えるように設計されており、統計的精度の高い解析が可能となっている。
一方、Pythonには基本的な統計解析ツールはあるが、Rほど豊富な高度な統計解析やモデリングパッケージが揃っているわけではない。特に、発現データやエピゲノムデータといった複雑なバイオデータ解析においては、Rの方が適していると言える。
2. バイオインフォマティクス特化のエコシステム
Bioconductorは、バイオインフォマティクスに特化した一貫性のあるエコシステムを提供しており、1000を超えるパッケージを利用できる。RNA-seq、ChIP-seq、ATAC-seq、シングルセルRNA-seq、メチル化解析、ネットワーク解析など、さまざまなバイオインフォマティクスの分野に対応したパッケージが揃っており、これらを統合して解析フローを構築できるのが大きな強みである。
Pythonはシーケンスデータの操作やAPI連携に強みを持つが、Bioconductorのように統合された解析フローをサポートするエコシステムとしては発展途上である。例えば、異なるオミクスデータの統合解析や、アノテーション情報を基にした高度な解析には、Bioconductorが提供する豊富なツール群が適している。
3. データの標準化と再利用性の高さ
Bioconductorは、データ構造や解析手法の標準化に優れており、例えば
SummarizedExperiment や SingleCellExperiment
などのデータ構造を用いることで、異なる解析ステップ間でのデータの互換性を保ちやすい。この統一されたデータ構造により、複数のパッケージ間でデータを効率的にやり取りしながら解析を進められる点が大きな強みである。
これに対して、Pythonでは標準化されたデータ構造が確立されておらず、異なるスクリプトやパッケージ間でデータを取り扱う際、ユーザーが手動で調整を行わなければならないことが多い。したがって、データの標準化と再利用性においても、Bioconductorの方が効率的である。
4. 可視化の柔軟性と高品質なグラフ生成
Rはデータ可視化においても非常に強力であり、ggplot2
をはじめとした高度な可視化ツールが豊富に揃っている。バイオインフォマティクスでは、解析結果を視覚的に解釈することが重要であり、ヒートマップ、ボルケーノプロット、PCAプロット、クラスタリングツリーなど、複雑なデータを効果的に表現できる点がRの強みである。
Pythonにも可視化ツールはあるが、Rの持つ柔軟性や高品質なグラフ生成能力には及ばない。特に、学術論文で求められるレベルの高品質な図表を簡単に生成できる点では、Rは他に類を見ないツールである。
5. 豊富なサポートとコミュニティ
Bioconductorは、充実したサポート体制と活発なコミュニティを持っている。豊富なドキュメントやチュートリアルが提供されており、初心者から上級者までが必要なサポートを受けやすい環境が整備されている。さらに、定期的に開催されるBioconductorカンファレンスなどを通じて、最新の技術や情報を学び、研究者同士が交流できる機会も多い。
Pythonにもコミュニティは存在するが、バイオインフォマティクス解析に特化した環境やサポートはRやBioconductorほど広範ではなく、解析手法の適用範囲も狭い。そのため、特にバイオインフォマティクスにおいてRを選択することが、学習や実践の面で有利である場合が多い。
Rを用いた下流解析で必要な操作スキルとは?
RおよびBiocondutorパッケージを用いて下流解析フローを適切に設計して実装するためには、大まかに分けて、二つの種類の本質的理解が必要となる。
各パッケージ関数の背後にある理論的な理解
各パッケージ関数を活用する際に生じる前後のデータ整理と変換・可視化
本講義ではRの基礎を扱うため、1については扱わない。簡単な補足をしておくと、各関数がどのようなアルゴリズムや統計手法に基づいているかを理解することは、信頼性の高い解析を行う上で不可欠である。例えば、RNA-seqにおけるlimmaやDESeq2のような発現変動解析を例にとると、正規化、分散のモデリング、仮説検定といった理論が背後に存在する。これらの理論的背景を理解していないと、関数のパラメータ設定や結果の解釈に誤りが生じる可能性がある。このような体系的知識は、各トピックごとに扱う方が具体的に扱えるだろう。
主に、2についてさらに考えていく。Bioconductorのパッケージには、Vignetteと呼ばれる使用例がMarkdown形式で添付されていることが多く、これを参考にすることで、解析フローを比較的簡単に実行することができる。初心者ユーザーでも、Vignetteに従うことで、関数に入力データを渡し、出力を次のステップに引き渡すといった基本的な操作が可能である。しかし、実際のデータはVignette通りの形式や条件で得られるとは限らない。全てを自分でコントロールできるわけではないからだ。むしろ例外的なケースに遭遇することの方が多く、ユーザーはその都度、Rの操作に習熟して対応する必要が出てくる。
では、これらの状況に対応するために、どのようなスキルが必要か?そのためには、Rでパッケージ関数を組み合わせて解析フローを構築する際に、ユーザーが行っている作業を理解することが重要である。具体的には、以下の操作が含まれる。
関数を正しく使う 目的に応じた関数を選び、適切に使う技術が必要。
関数に対する入力データと引数を用意する 関数が要求する入力データやパラメータを整形・設定する必要がある。
関数からの出力データを整理・可視化する 出力を整形し、次のステップに入力したり、可視化・解釈できる形式に変換する。
Rでの解析における「親切な関数」と「不親切な関数」
Rのパッケージ関数には、ユーザーにあまり手間をかけずに動作する「親切な関数」と、詳細な設定や準備が必要な「不親切な関数」が存在する。親切な関数は、入力データがある程度整っていれば、自動的に多くの処理を行ってくれる。一方で、不親切な関数は、データの整形や前処理をユーザーに要求する。ユーザーがどのようにデータを整形し、必要な変換を行うかが、解析のスムーズさに大きく影響する。
親切な関数:本来の処理以外も色々と想定して自動的にやってくれる
不親切な関数:本来の処理以外はやらず、ユーザーに任せる
不親切な関数という表現に驚くかもしれないが、開発者の立場からすると、親切な関数を作ることは非常に難しい。なぜなら、全く未知のユーザーを想定して、あらゆる可能性に対応するように作るのは膨大な作業量を必要とするからである。実際、ユーザーのスキルやニーズは多様であり、どのように関数を使うかも予測しづらい。このため、ユーザー自身が必要な操作を行うよう促されるのは仕方ないことかもしれない。さらに、オープンソースで提供されている関数の場合、善意で公開されていることから、その点を批判するのは必ずしも適切ではないかもしれない。また、汎用性を高めるために、意図的にユーザーに自由な操作を許す設計がなされている場合もある。
関数の出力データの分析・整理
また、パッケージ関数の出力から必要な情報を取り出すためには、ユーザー自身がフィルター条件を設定し、出力データを分析する必要がある。さらに、パッケージには出力データを可視化するための関数が用意されていることも多いが、その多くは見栄えが悪く、最近の洗練されたビジュアルに比べて満足できない場合がある(これも前述の「不親切な関数」と同じ理由による)。そのため、カスタマイズの必要性から、別の可視化パッケージを使用する選択肢が出てくることもある。この場合も、ユーザーはその可視化のために出力データを整形・変換する必要がある。
必要なスキルを習得するためのステップ
このような理由から、ユーザーはRにある程度習熟し、関数を使いこなすために自ら処理を行う必要が生じる。以下に、RNA-seqの発現変動解析を例に取り、各ステップでユーザーがどの操作を行うべきかを示す。これにより、データ構造の理解やベクトル操作、データの整形・可視化の重要性が明確になる。
例1:RNA-seq 発現変動解析 (limma)
RNA-seqデータの発現変動解析のフローを示し、どの部分が自動的に処理され、どこでユーザーの介入が必要かを解説する。
# 1. データの読み込みとDGEListオブジェクトの作成
# ユーザーが行う部分
library(limma)
library(edgeR)
# カウントデータとサンプル情報の読み込み
count_data <- read.table("counts_matrix.txt", header=TRUE, row.names=1)
meta_data <- read.csv("metadata.csv", row.names=1)
# DGEListオブジェクトの作成
dge <- DGEList(counts=count_data, samples=meta_data)
# 2. アノテーションデータの統合
# ユーザーが行う部分
annotation_data <- read.table("gene_annotation.txt", header=TRUE, row.names=1)
dge$genes <- annotation_data[rownames(dge), ]
# 3. 正規化
# edgeRが自動的に行う部分
dge <- calcNormFactors(dge)
# 4. メタ情報を整理してデザインマトリクスを作成
# ユーザーが行う部分
group <- factor(meta_data$condition)
design <- model.matrix(~0 + group)
colnames(design) <- levels(group)
# 5. 遺伝子のフィルタリング
# ユーザーが行う部分
keep <- filterByExpr(dge, design)
dge <- dge[keep, , keep.lib.sizes=FALSE]
# 6. 分散縮小モデルの適用
# voomが行う部分
v <- voom(dge, design, plot=TRUE)
# 7. フィッティングと経験ベイズによる発現変動解析
# limmaが自動的に行う部分
fit <- lmFit(v, design)
# 8. コントラストの設定と解析実行
# ユーザーが行う部分
contrast <- makeContrasts(Treatment-Control, levels=design)
fit2 <- contrasts.fit(fit, contrast)
fit2 <- eBayes(fit2)
# 9. 結果の整形と出力
# ユーザーが行う部分
results <- topTable(fit2, adjust="fdr", number=Inf)
# p値やFoldChangeに基づいてフィルタリング
significant_genes <- results[results$adj.P.Val < 0.05 & abs(results$logFC) > 2, ]
write.csv(significant_genes, file="limma_differential_expression_results.csv")各ステップの説明
1. DGEListオブジェクトの作成
- ユーザーの操作:
カウントデータとサンプル情報を読み込み、
DGEListオブジェクトにまとめる。- 必要なスキル: データの入出力操作、データ構造の理解。
2. アノテーションデータの統合
- ユーザーの操作:
外部のアノテーションデータを読み込み、
DGEListオブジェクトに統合する。- 必要なスキル: データのマージや整形操作、アノテーションデータの管理。
3. 正規化
- 自動処理:
calcNormFactors()関数がサンプル間のライブラリサイズを補正する。- 必要なスキル: 自動的に処理されるが、正規化の理論的背景を理解しておくことが重要。
4. メタ情報の整理とデザインマトリクスの作成
- ユーザーの操作:
サンプル条件に基づき、比較するためのデザインマトリクスを作成する。
- 必要なスキル: サンプル情報の整理、デザインマトリクスの作成、ベクトル操作。
5. 遺伝子のフィルタリング
- ユーザーの操作:
filterByExpr()関数を使って、解析対象となる遺伝子をフィルタリングする。- 必要なスキル: 適切なフィルタリング条件の設定、データのスクリーニング。
6. 分散縮小モデルの適用
- 自動処理:
voom()関数が、RNA-seqデータの分散と平均の関係をモデル化し、正規化された分散縮小モデルを適用する。- 必要なスキル: モデルの適用に伴う理論の理解(自動処理ではあるが、背後にある統計的概念の理解が求められる)。
7. フィッティングと経験ベイズによる発現変動解析
- 自動処理:
lmFit()関数で線形モデルをフィッティングし、contrasts.fit()関数でコントラストを適用、eBayes()で経験ベイズによる解析を行う。- 必要なスキル: 統計モデルの理解、フィッティング手法の知識。
8. コントラストの設定と解析実行
- ユーザーの操作:
makeContrasts()関数を使用して、比較したい条件(例: Treatment vs. Control)を指定する。- 必要なスキル: 比較条件の設定、統計解析に必要なモデル構築。
9. 結果の整形と出力
- ユーザーの操作:
topTable()で発現変動遺伝子の結果を抽出し、フィルタリング基準に従って重要な遺伝子を選別する。- 必要なスキル: 結果の整形、可視化、出力の整理。
習得すべきRスキル
上の例題におけるR操作の本質的要素を抽出してまとめると以下のようになろう。
- データ構造の理解:
DGEListオブジェクトやlimmaのデータ構造を理解することで、解析の流れを効率的に進めることができる。 - ベクトル操作の習熟:
rownames()やcolnames()などを使ったメタ情報やアノテーションの整形、データのフィルタリング操作に習熟する。 - 結合操作: メタ情報やアノテーション情報を解析データに結合し、解析の準備を整える操作を習得する。
- 可視化の習熟: 発現変動遺伝子を可視化し、結果を直感的に解釈できるようにする技術を学ぶ。
定量アッセイに対する下流解析の一般的フローと必要なR操作
定量アッセイに対する下流解析の流れには、どのアッセイ系であっても一定の「型」や「モデル」が存在する。これは、アッセイ系が多様であっても、ハイスループット測定の原理が基本的にシークエンサーや質量分析(かつてはマイクロアレイ)のいずれかに依存しており、それぞれに共通する部分があるためである。バイオインフォマティクスにおいて、データ解析手法の本質や原理を理解する上で、このメタ的な視点は極めて重要である。つまり、測定限界やアーチファクト、ノイズ構造の原理は多くの場合、異なる機器間で共通しており、見た目が異なる部分も、数理的な方法としては大きく変わらないことが多い。メタ的視点でデータ解析の流れを「モデル」として理解しておくと、アッセイ系ごとの特有な処理があっても、解析全体の見通しを立てやすくなる。
以下に、定量アッセイ全般に共通するデータ準備、前処理、下流解析の主なプロセスをまとめる。
1. データの統合とアノテーション
多くの定量アッセイでは、複数の実験や異なるデータセットを統合する必要がある。例えば、RNA-seqやプロテオミクスデータの統合において、異なるサンプルや異なる実験条件間で一貫性を持たせるために、以下のようなステップが含まれる。
- データセットの統合
- 異なるバッチやサンプルから取得したデータを統合する。Rの
merge()やdplyrパッケージのleft_join()を使用して、共通のキー(遺伝子IDやタンパク質IDなど)に基づき、複数のデータを統合する。
- 異なるバッチやサンプルから取得したデータを統合する。Rの
- アノテーションの追加
- 遺伝子やタンパク質のIDに基づいて、外部のアノテーションデータ(例えば、HGNCやEnsemblデータベース)から遺伝子名や機能的アノテーションを取得する。Rの
biomaRtパッケージやAnnotationDbiを用いることで、アノテーションを効率よく追加することができる。
- 遺伝子やタンパク質のIDに基づいて、外部のアノテーションデータ(例えば、HGNCやEnsemblデータベース)から遺伝子名や機能的アノテーションを取得する。Rの
- メタ情報の処理
- サンプルや条件に関するメタ情報(性別、年齢、実験条件など)を統合し、解析に反映させる。
SummarizedExperimentやSingleCellExperimentのcolData()関数を使ってメタ情報を統合し、解析に活用する。
- サンプルや条件に関するメタ情報(性別、年齢、実験条件など)を統合し、解析に反映させる。
2. データの前処理
データの正規化やフィルタリング、バッチ効果の除去など、前処理はデータの品質を保ち、解析結果を信頼できるものにするために不可欠なステップである。
- データの正規化とスケーリング
- 異なるサンプル間でのバッチ効果や測定バイアスを取り除くために、定量データを正規化する。
DESeq2のrlog()やlimmaのvoom()などを使用して、スケーリングとバッチ効果の補正が行われる。
- 異なるサンプル間でのバッチ効果や測定バイアスを取り除くために、定量データを正規化する。
- フィルタリング
- サンプルの低品質データやノイズを取り除くために、各特徴(遺伝子やタンパク質)の総和や分散に基づいてフィルタリングを行う。
apply()関数を用いて遺伝子ごとの分散を計算し、一定の基準を満たさない遺伝子を除外する。
- サンプルの低品質データやノイズを取り除くために、各特徴(遺伝子やタンパク質)の総和や分散に基づいてフィルタリングを行う。
- バッチ効果の補正
- 異なるバッチ間のシステム的なバイアスを補正するために、
ComBatやlimmaのremoveBatchEffect()を使ってバッチ効果を除去する。
- 異なるバッチ間のシステム的なバイアスを補正するために、
3. 次元削減とクラスタリング
次元削減やクラスタリングは、データの構造を可視化し、サンプル間の関係性やパターンを見つけるために重要なステップである。
- 次元削減
- 高次元データを2次元または3次元に圧縮し、データの全体的なパターンを視覚化する。主成分分析(PCA)、t-SNE、UMAPといった手法が一般的で、Rでは
Seuratやggplot2を使用して次元削減結果を可視化する。
- 高次元データを2次元または3次元に圧縮し、データの全体的なパターンを視覚化する。主成分分析(PCA)、t-SNE、UMAPといった手法が一般的で、Rでは
- クラスタリング
- サンプルや条件をクラスタリングし、類似したパターンを持つグループを特定する。K-meansや階層的クラスタリング、
Seuratによるシングルセルデータのクラスタリングなどが広く利用される。
- サンプルや条件をクラスタリングし、類似したパターンを持つグループを特定する。K-meansや階層的クラスタリング、
4. 機械学習と予測モデル
次に、データの学習と予測に進む段階では、機械学習アルゴリズムが活用される。RではcaretやrandomForestなどのパッケージを使用して、モデルの構築や予測精度の評価が行われる。
- モデルの訓練とテスト
- 学習データに基づいて予測モデルを構築し、交差検証によってモデルの性能を評価する。
caretやglmnetを使用して、複数のアルゴリズムを試し、最適なモデルを選択する。
- 学習データに基づいて予測モデルを構築し、交差検証によってモデルの性能を評価する。
- 特徴選択
- 高次元データの特徴量を削減し、重要な特徴を選択する。例えば、遺伝子発現データでは、発現が変動している遺伝子を選別してモデルに使用することが一般的である。
5. 発現変動解析と機能的アノテーション
データの特徴や条件間の違いを調べ、発現変動解析を行う。DESeq2やlimmaなどのパッケージを使用して、発現が変動している特徴(遺伝子やタンパク質)を特定する。
- 発現変動解析
- 異なる条件間での遺伝子やタンパク質の発現量の違いを調べ、統計的に有意な特徴を特定する。
DESeq2やedgeRなどが一般的に使用される。
- 異なる条件間での遺伝子やタンパク質の発現量の違いを調べ、統計的に有意な特徴を特定する。
- パスウェイ解析や機能的アノテーション
- 発現変動した遺伝子やタンパク質が関与するパスウェイや機能を明らかにするために、
clusterProfilerやReactomePAを使ってアノテーションを行う。
- 発現変動した遺伝子やタンパク質が関与するパスウェイや機能を明らかにするために、
6. ネットワーク解析
- 遺伝子やタンパク質の相互作用解析
- 得られた特徴の相互作用ネットワークを構築し、遺伝子やタンパク質の重要な相互作用を明らかにする。Rでは、
WGCNAやigraphを使用してネットワーク解析が行われる。
- 得られた特徴の相互作用ネットワークを構築し、遺伝子やタンパク質の重要な相互作用を明らかにする。Rでは、
Rでデータを自在に操るための基礎
上記のデータ解析の「型」のうち、ユーザーが自ら行わなければならない本質的な操作に着目して抽出すると、データの前処理や整形、出力の可視化など多くのステップなど見えてくる。以下は、そのような関数を利用する際にユーザーが習熟すべき主要なスキルをまとめたものである。
1. データの整形と前処理
パッケージ関数がデータの整形や前処理を自動で行わない場合、ユーザーは自らデータを適切な形式に変換する必要がある。
- データのインポートとフォーマットの調整
- データを適切な形式でインポートし、欠損値の処理や列のリネームなどを行うスキル。
read.csv(),fread(),data.frame(),as.factor()などの基本操作。
- データのクレンジング
- 欠損値や異常値を検出・修正し、データの質を確保する。
is.na(),na.omit(),dplyr::mutate()などを使ってデータをクレンジングするスキル。
2. データのフィルタリングとサブセット化
関数がすべてのデータを自動的に処理しない場合、ユーザーは必要な部分を選択して分析する必要がある。
- データのサブセット化
- 必要なサンプルや変数だけを抽出し、解析用にデータをサブセット化するスキル。
subset(),filter(),select()を用いた操作。
- フィルタリング
- 基準に基づいてデータをフィルタリングし、解析に適さない部分を除外する。
apply(),rowMeans(),filterByExpr()などで遺伝子やタンパク質のフィルタリング。
3. データのマージとアノテーション
不親切な関数はしばしば、データのアノテーションや外部データの統合を自動的に行わない。ユーザーは自ら必要な情報を統合する必要がある。
- データセットのマージ
- 異なるソースのデータセットを適切に統合し、共通キー(遺伝子IDやサンプルID)に基づいて結合するスキル。
merge(),dplyr::left_join()などを使ったデータのマージ。
- アノテーションの追加
- 遺伝子やタンパク質に関するアノテーションデータを外部のデータベース(
biomaRtやAnnotationDbi)から取得し、統合する操作。
- 遺伝子やタンパク質に関するアノテーションデータを外部のデータベース(
4. 関数の出力結果の整形と解析
パッケージ関数が自動的に出力を整形しない場合、ユーザーはその結果を適切に処理・可視化する必要がある。
- 出力データの整形
- 関数の出力データがリストや複雑な構造である場合、必要な部分を抽出し、解析や可視化に使える形式に整形する。
unlist(),lapply(),sapply()などの関数でリスト構造のデータを操作。
- 結果のフィルタリング
- 統計的に有意な結果(例えばFDRやp値に基づく)を抽出し、重要な結果を報告する。
topTable(),subset()で結果をフィルタリングし、適切な基準で解析結果を選別する。
5. カスタムの可視化
関数が結果を可視化する際、標準的な出力では見栄えが悪い場合が多い。このため、ユーザーはカスタムで可視化を行うスキルが求められる。
- カスタムプロットの作成
- 基本的なプロット機能ではなく、
ggplot2やplotlyを使ってカスタマイズしたプロットを作成するスキル。 - グラフの軸、凡例、タイトルなどを自由にカスタマイズして、わかりやすいビジュアルを作成する。
- 基本的なプロット機能ではなく、
- 結果の可視化
- クラスタリングや次元削減結果、発現変動解析の結果を可視化して、データのパターンや有意性を視覚的に表現する。
- PCA, t-SNE, UMAPの結果をプロットするための技術
(
ggplot2,Seuratなど)。
6. パイプラインの構築と自動化
関数の連続的な適用が必要な場合、パイプラインを構築して効率化するスキルが重要。
- データ処理のパイプライン化
%>%(パイプオペレーター)を使った一連の操作を効率化する。dplyrやtidyrを使い、複数の操作を連続して実行するスキル。
- スクリプトの自動化
- 同じ解析を繰り返す場合、スクリプトを作成して処理を自動化するスキル。
forループやapplyファミリーを使って、操作を繰り返し処理する。
パッケージ関数の活用を前提としたR操作の本質的な基礎とは?
では、上記の操作スキルにはまだ、Rの操作として重複している部分がある。それらをさらにまとめていくと、下流解析において、パッケージ関数の利用を前提としたRの基礎とは、おそらく以下の点に集約される。
- 入力・出力データのファイル形式およびデータ形式の多様性
- 生物学的データ解析では、さまざまなデータ形式やファイル形式(CSV、BAM、FASTQ、VCFなど)を取り扱う。ユーザーは、Rの
read.csvやread.table、RsamtoolsなどのBioconductorパッケージを使用してこれらのファイルを適切に読み込む必要がある。また、データセットごとに異なるフォーマットを統一するために、データの変換や統合を行う操作スキルが必要である。ファイル形式の違いによって、読み込み時に適切な設定や前処理が必要になるため、これらの操作はR操作の基本スキルに含まれる。
- 生物学的データ解析では、さまざまなデータ形式やファイル形式(CSV、BAM、FASTQ、VCFなど)を取り扱う。ユーザーは、Rの
- Rにおけるデータ構造の理解と操作スキル
- Bioconductorパッケージを使った解析では、
SummarizedExperiment、SingleCellExperiment、ExpressionSetといった特殊なS4クラスのデータ構造を取り扱う。これらの構造に慣れるためには、通常のdata.frameやmatrixに対する基本操作の理解が不可欠である。ユーザーは、基本的なデータフレーム操作に加えて、S4オブジェクトのメタ情報(例:colDataやrowData)を抽出したり、アッセイデータを取り扱う際の操作に習熟する必要がある。これらのデータ構造に対する基本的な理解が、データ解析の前提となる。
- Bioconductorパッケージを使った解析では、
- データの整形とサマリー作成
- Bioconductorパッケージの関数を使う前に、データを適切に整形する必要がある。遺伝子ごとの発現データやメタデータをフィルタリング・サブセット化し、解析に適した形に変換することが求められる。また発現変動解析を行う際には、入力データを正規化したり、遺伝子やサンプルごとのフィルタリングを行う必要があり、これらの操作にはRの基本的な関数(
applyやfilterなど)を用いる。データの変換やサブセット化は、解析結果の品質に大きく影響するため、これも重要なスキルである。
- Bioconductorパッケージの関数を使う前に、データを適切に整形する必要がある。遺伝子ごとの発現データやメタデータをフィルタリング・サブセット化し、解析に適した形に変換することが求められる。また発現変動解析を行う際には、入力データを正規化したり、遺伝子やサンプルごとのフィルタリングを行う必要があり、これらの操作にはRの基本的な関数(
- 可視化のためのデータ整形と統計的要約
- 多くの解析結果は、最終的に可視化して解釈される。例えば、主成分分析(PCA)やクラスタリング解析の結果を適切に可視化するために、データを適切に整形・要約する必要がある。
ggplot2を用いた可視化では、ロング形式(long format)のデータに変換したり、必要な統計的要約を行うことが求められる。適切な可視化を行うためには、データを整形するスキルが欠かせない。また、プロットのカスタマイズやデータのサブセット化も、R操作における基本的な部分を占めている。
- 多くの解析結果は、最終的に可視化して解釈される。例えば、主成分分析(PCA)やクラスタリング解析の結果を適切に可視化するために、データを適切に整形・要約する必要がある。
これらの4つの点は、Bioconductorパッケージ関数を活用する際の前提となる基礎的なR操作に該当する。パッケージ関数が提供する自動処理部分と、ユーザーが手動で行うデータ準備や変換・整形部分の間にある「操作の溝」を埋めるために、これらのRスキルが不可欠となる。また、これらは解析の柔軟性を高め、問題が発生した際のトラブルシューティングにも役立つと思われる。
データのライフサイクルを理解する
Rにおけるデータ操作を自在に行うためには、データのライフサイクルを把握することが基本となる。バイオインフォマティクス解析において、データの流れは以下のサイクルに基づいて進行する。
- データの読み込み: CSV、Excel、FASTQ、BAMなど、多様な形式のデータをRに取り込む。
- 前処理: データのクリーニング、欠損値の補完、データの正規化など、解析の前提条件を整える。
- モデリングと分析: 統計モデルや機械学習を用いて、データの構造を探り、予測や分類を行う。
- 要約と可視化: 分析結果を要約し、視覚的にデータを理解するためのグラフやプロットを作成する。
- レポート作成と結果の保存: 解析の結果をまとめ、再現性のある形で記録・保存する。
このライフサイクルは一方向のプロセスではなく、前処理と分析、可視化を何度も繰り返しながら最適化を進めていくことが多い。
バイオインフォマティクス解析におけるデータの多様性
バイオインフォマティクス解析では、扱うデータの形式が非常に多様である。一般的な表形式データだけでなく、配列データやゲノムアノテーションデータなど、独自の形式で保存されたデータを適切に読み込むための知識が不可欠である。たとえば、次のようなデータ形式が存在する。
| データ形式 | 説明 | 形式例 | Rパッケージ | 読み込み関数 | データ構造 | 書き込み関数 | 使用頻度 |
|---|---|---|---|---|---|---|---|
| CSV/TSV | カンマまたはタブ区切りのテーブル形式データ | sample.csv, sample.tsv |
base R, data.table |
read.csv(), fread() |
data.frame | write.csv(), fwrite() |
高 |
| Excel (XLS, XLSX) | Excelスプレッドシート | sample.xlsx, sample.xls |
readxl, openxlsx |
read_excel(), read.xlsx() |
data.frame | write.xlsx() |
中 |
| JSON | JavaScript Object Notation。構造化データ | sample.json |
jsonlite, rjson |
fromJSON(), toJSON() |
list/data.frame | write_json() |
中 |
| YAML | 構造化データ(YAML形式) | config.yaml |
yaml |
yaml.load(), yaml.load_file() |
list/data.frame | write_yaml() |
低 |
| XML | 階層型データ | data.xml |
XML, xml2 |
xmlParse(), read_xml() |
list/data.frame | write_xml() |
低 |
| HDF5 | 階層型データフォーマット | sample.h5, sample.hdf5 |
rhdf5 |
h5read(), h5ls() |
list/array | h5write() |
中 |
| RDS | Rオブジェクト保存形式 | object.rds |
base R |
readRDS() |
R object | saveRDS() |
高 |
| RData | 複数のRオブジェクト保存形式 | data.RData |
base R |
load() |
environment | save() |
中 |
| Matrix Market (MTX) | スパース行列形式 | matrix.mtx, matrix.mtx.gz |
Matrix, Matrix.utils |
readMM() |
Matrix/Matrix-like | writeMM() |
中 |
| FASTQ | DNA/RNA配列リードシーケンスデータ | sample.fastq, sample.fastq.gz |
ShortRead, Biostrings |
readFastq(), readDNAStringSet() |
ShortReadQ | writeFastq() |
高 |
| FASTA | 配列データフォーマット | sample.fasta, sample.fa |
Biostrings |
readDNAStringSet(), readAAStringSet() |
DNAStringSet | writeXStringSet() |
高 |
| SAM/BAM | シーケンスアライメントデータ | sample.sam, sample.bam |
Rsamtools |
scanBam(), readBamFile() |
BamFile/BamFileList | writeBamFile() |
高 |
| VCF | バリアントコールフォーマット | variants.vcf, variants.vcf.gz |
VariantAnnotation |
readVcf() |
VCF | writeVcf() |
中 |
| GFF/GTF | ゲノムアノテーションフォーマット | annotation.gff, annotation.gtf |
rtracklayer |
import() |
GRanges | export() |
中 |
| BED | ゲノム領域データ | regions.bed |
rtracklayer, GenomicRanges |
import() |
GRanges | export() |
中 |
| Count Matrix | 遺伝子発現量のカウントデータ | counts.txt, counts_matrix.tsv |
edgeR, DESeq2 |
read.table(), read.delim() |
matrix/data.frame | write.table(), write.delim() |
高 |
| mzML/mzXML | 質量分析のデータ | sample.mzML, sample.mzXML |
mzR, MSnbase |
openMSfile(), readMSData() |
MSnSet | writeMSData() |
中 |
| BigWig | ゲノムカバレッジデータ | coverage.bw |
rtracklayer |
import.bw() |
GRanges | export.bw() |
中 |
| AnnData (H5AD) | シングルセルデータ用HDF5形式 | sample.h5ad |
zellkonverter |
readH5AD() |
SingleCellExperiment | writeH5AD() |
中 |
| SQLite | SQLデータベース形式 | database.sqlite |
DBI, RSQLite |
dbConnect(), dbReadTable() |
SQLiteConnection | dbWriteTable() |
低 |
| Feather | 高速読み込み用バイナリ形式 | data.feather |
feather, arrow |
read_feather(), write_feather() |
data.frame | write_feather() |
低 |
| Parquet | 高速読み込み用列指向フォーマット | data.parquet |
arrow |
read_parquet(), write_parquet() |
data.frame | write_parquet() |
中 |
| GZ/BZ2 | 圧縮形式(gzip/bzip2)で保存されたファイル | data.gz, data.bz2 |
R.utils, base R |
gunzip(), gzfile(),
bunzip2() |
CompressedFile/Raw | gzip(), bzfile() |
高 |
| RLE | ランレングスエンコーディング形式 | data.rle |
Rle |
runLength() |
Rle | N/A | 低 |
| GMT | 遺伝子セット定義フォーマット | geneset.gmt |
GSEABase, fgsea |
getGmt(), gmtPathways() |
GeneSetCollection | N/A | 中 |
| SIF | ネットワーク相互作用データ形式 | network.sif |
igraph |
read.graph() |
igraph/network | write.graph() |
低 |
| TOML | 構造化設定ファイルフォーマット | config.toml |
toml |
toml::from_toml() |
list/data.frame | toml::to_toml() |
低 |
データ読み込みの基盤スキル
データ読み込みのスキルの本質は、想定するデータ形式に沿わない場合に、適切な引数や関数のオプションを活用し、データを正確に読み込むための試行錯誤ができるスキルにある。バイオインフォマティクスにおけるデータは非常に多様であり、必ずしも整った形式で提供されるとは限らない。そのため、以下のような能力が特に重要である。
1. データ形式の理解
さまざまなファイル形式(CSV, TSV, FASTQ, VCF, GTF, GMT, SIFなど)に対応し、それぞれの形式の特徴を理解することが第一歩である。例えば、列の区切り文字、ヘッダーの有無、改行コードの違いなど、データ形式の違いが読み込みに与える影響を理解する必要がある。
2. 引数やオプションの柔軟な使用
各読み込み関数(fread, read.table,
read.csv, read.delim
など)には、データのフォーマットに合わせて引数を調整する機能が豊富に用意されている。これらを活用して、データ形式に応じた適切な読み込みを行うことがスキルの中心である。
区切り文字の指定 (
sep,delimiter): 区切り文字が予期せず異なる場合(例: カンマ区切り vs. タブ区切り)、引数を使って明示的に指定する。fread("data.txt", sep = "\t") # タブ区切りの場合ヘッダーの有無 (
header): ファイルの先頭にヘッダーがない場合や、複数行のヘッダーがある場合、header = FALSEやskip引数を使う。fread("data.txt", header = FALSE, skip = 2) # ヘッダーが2行分ある場合データ型の強制変換 (
colClasses,as.is): 特定の列が予期せぬデータ型(例: 数値が文字列として扱われる)として読み込まれた場合、colClasses引数を用いて強制的に型を指定する。fread("data.txt", colClasses = c("GeneID" = "character", "Expression" = "numeric"))欠損値の処理 (
na.strings): 欠損値が異なる表現(例: “NA”, “-”, “null” など)で表記されている場合、それらをna.stringsで明示して読み込み時に処理する。fread("data.txt", na.strings = c("NA", "-", "null"))
3. 試行錯誤とデータの確認
読み込み後のデータが期待通りであるかどうかを確認し、必要に応じて再調整するプロセスが重要である。データの一部を確認するために、head()
や str()
などの関数を用い、誤って読み込まれた箇所があれば再度引数を調整して試行錯誤する。
# データの構造を確認
str(quant_data)
head(quant_data)4. 想定外のデータフォーマットへの対応力
時には、データが標準的な形式ではなく、不規則な改行や複数の区切り文字が混在している場合もある。このような場合、読み込み関数だけで対処できないことも多く、テキスト処理やファイルを前処理する技術が求められる。
正規表現を用いた前処理: テキストファイルが一貫性を欠いている場合、
sedやawkなどのコマンドラインツールや、Rのstringrパッケージで前処理を行う。library(stringr) data <- readLines("data.txt") data_clean <- str_replace_all(data, "[^[:alnum:]\t]", "") # 不要な記号を削除
5. データがどうしても読み込めない場合の対応
データが標準的な関数ではどうしても読み込めない場合、データを一旦
readLines や scan
で読み込み、手作業でクリーニングすることがある。このような手法を使うことで、複雑なデータ構造に対応できる。
# readLinesでファイルを行単位で読み込む
raw_data <- readLines("data.txt")
# 不要な行や空行を削除
raw_data_clean <- raw_data[raw_data != "" & !grepl("^#", raw_data)] # 空行やコメント行を削除
# データを適切な形式に変換
data_matrix <- read.delim(textConnection(raw_data_clean), sep = "\t")6. シェルスクリプトによる事前処理
Rだけでなく、シェルスクリプトを用いたデータ整形も有効である。特に大規模なデータセットに対しては、シェルで事前に処理することで効率化できる。シェルを使った基本的な整形操作を以下に示す。
1. head でデータの確認
head data.txt # ファイルの先頭10行を確認2. sed や awk を用いたデータ整形
- 区切り文字の変換(例: カンマをタブに置換)
sed 's/,/\t/g' data.txt > cleaned_data.txt- 不要な行の削除(例: 空行や特定のパターンを含む行を削除)
sed '/^$/d' data.txt > cleaned_data.txt # 空行を削除
awk '!/^#/' data.txt > cleaned_data.txt # コメント行を削除- 列の抜き出し(例: 2列目と4列目を抽出)
cut -f2,4 data.txt > selected_columns.txt3. grep で特定行を抽出
grep "Pattern" data.txt > filtered_data.txt7. データ品質チェックとクリーニング
最後に、読み込んだデータが解析に適しているかどうかの品質チェックも不可欠である。読み込み時のエラーや警告を無視せず、データを正しくクリーニングするプロセスを踏むことで、後続の解析をスムーズに進められる。
データ読み込みのスキルは、単に関数を使うだけでなく、データ形式の多様性に柔軟に対応し、問題に応じて引数を調整して試行錯誤する力にある。特に、バイオインフォマティクスにおいては、データ形式の多様さと複雑さに応じて適切な処理を行い、シェルスクリプトなども活用して柔軟に対応することが求められる。これらのスキルは、データ解析の最初のステップとして極めて重要であり、正確なデータの読み込みが解析結果の信頼性を高める基盤となる。
データ読み込みスキルの演習例題
以下に、データ読み込みのスキルを養うための例題をいくつか示す。これらの演習は、異なる形式のデータを読み込み、適切に整形するための技術を実践的に学ぶことが目的である。
例題1: CSVファイルの読み込みと整形
課題:
次のようなCSVファイル gene_expression.csv
を読み込んでください。このデータには、ヘッダー行が2行存在し、カンマ区切りで遺伝子発現量データが格納されています。1行目はファイルの説明、2行目がヘッダーです。空白セルはNAとして扱ってください。
gene_expression.csv:
# Gene expression data from experiment XYZ
GeneID,SampleA,SampleB,SampleC
ENSG000001,10.5,9.8,,
ENSG000002,7.5,NA,12.3
ENSG000003,5.0,4.8,5.1演習:
- ファイルを読み込み、ヘッダー行をスキップして2行目からデータを扱うように設定する。
- 欠損値(空白セル)を
NAとして処理する。 GeneID列を文字列型に指定し、数値列の型を確認する。- データの一部を表示し、正しく読み込めたかを確認する。
解答例:
# CSVファイルの読み込み
gene_data <- fread("gene_expression.csv", skip = 1, na.strings = c("", "NA"))
# GeneID列を文字列型にする
gene_data[, GeneID := as.character(GeneID)]
# データの確認
str(gene_data)
head(gene_data)例題2: タブ区切りファイルの読み込みと前処理
課題:
次のようなタブ区切りファイル sample_metadata.txt
には、実験に関するメタデータが格納されています。このデータにはいくつかの行がコメント(#で始まる行)として記載されており、空白行も含まれています。このファイルを読み込み、不要なコメント行や空白行を削除してください。
sample_metadata.txt:
# Sample metadata from RNA-seq experiment
SampleID Condition Replicate
# Comment about the experiment
SampleA Control 1
SampleB Control 2
SampleC Treatment 1
SampleD Treatment 2演習:
- コメント行(
#で始まる行)と空白行を削除し、データを読み込む。 - 各列のデータ型を確認し、正しく読み込めているか確認する。
解答例:
# ファイルを行単位で読み込む
raw_metadata <- readLines("sample_metadata.txt")
# 空白行とコメント行を削除
clean_metadata <- raw_metadata[raw_metadata != "" & !grepl("^#", raw_metadata)]
# データをタブ区切りで読み込む
metadata <- read.delim(textConnection(clean_metadata), sep = "\t")
# データの確認
str(metadata)例題3: FASTQファイルの読み込みとクリーニング
課題:
次のようなFASTQ形式のファイル reads.fastq
には、シークエンスリードデータが含まれています。このファイルには改行が不規則な部分が含まれているため、まずはテキストとして読み込み、クリーニングする必要があります。最終的に、各リードが4行ごとに正しく整列されているかを確認してください。
reads.fastq:
@SEQ_ID1
GATTTGGGG
+
!!!!!
@SEQ_ID2
GATCAAGGA
+
!!!!!
@
SEQ_ID3
GATTAGGAA
+
!!!!!演習:
readLines()を使ってファイルを読み込み、リードの欠けている行を手作業で修正する。- 各リードが4行ごとに正しい形式で並んでいるかを確認する。
解答例:
# FASTQファイルを読み込む
fastq_data <- readLines("reads.fastq")
# 不規則な改行を修正する
fastq_data_clean <- fastq_data[fastq_data != ""] # 空行を削除
# リードが4行ごとに正しく整列されているか確認
n_lines <- length(fastq_data_clean)
if (n_lines %% 4 == 0) {
print("リードが正しく整列しています")
} else {
print("リードの整列に問題があります")
}
# データを表示
print(fastq_data_clean)例題4: CSVファイルの事前チェックとシェルスクリプトによる整形
課題:
次のような大規模なCSVファイル large_data.csv
を読み込む必要があります。しかし、ファイルが非常に大きいため、まずシェルスクリプトを使ってファイルの先頭を確認し、不要な列を事前に削除したいと考えています。以下のステップに従って処理を進めてください。
large_data.csv:
SampleID,GeneA,GeneB,GeneC,GeneD,Metadata
1,5.4,3.2,7.1,2.4,SampleA_Info
2,6.1,2.8,6.9,3.3,SampleB_Info
3,5.9,3.5,7.4,2.9,SampleC_Info
...演習:
- シェルコマンドを使ってファイルの先頭を確認し、不要な列(
Metadata列)を削除する。 - 整形されたファイルをRで読み込み、データの一部を確認する。
解答例:
シェルでの整形操作(Linux環境の場合)
# ファイルの先頭10行を確認
head large_data.csv
# Metadata列を削除して新しいファイルに保存
cut -d, -f1-5 large_data.csv > cleaned_large_data.csvRでの読み込み
# 整形されたCSVファイルを読み込む
large_data <- fread("cleaned_large_data.csv")
# データの確認
str(large_data)
head(large_data)オブジェクトの構造を理解する重要性
Rにデータを読み込むと、そのデータはオブジェクトとして保存される(表中の「データ構造」を参照)。ここで、オブジェクトの構造を正しく理解しないと、適切なデータ操作や解析が行えない。なぜならRの関数は、特定のオブジェクト構造(ベクトル、行列、データフレーム、リストなど)を前提として動作するため、入力値や出力値の構造に誤りがあると、エラーが発生する。特に、バイオインフォマティクス解析では、SummarizedExperiment、GRanges、SingleCellExperiment
など、複雑な情報を統合的に管理する高度なデータ構造が頻繁に使われる。これらのオブジェクトを適切に操作するためには、構造を理解し、各ステップに応じた操作を選択する能力が必要である。
オブジェクト間の変換と柔軟なデータ操作
解析を進める上で、データ構造を別の形式に変換する必要が生じることがある。たとえば、データを行列形式で保持しつつ、解析結果をデータフレームに変換して可視化を行うなど、柔軟なデータ操作が求められる。Rには、as.data.frame()
や as.matrix()
などの関数があり、データの形式を適切に変換することが可能である。これにより、異なるデータソースの統合や解析結果の変換が容易になり、解析の効率が向上する。
継続的な試行錯誤とデータ解析の最適化
データのライフサイクルとオブジェクトの構造を理解した上で、Rによるデータ操作は一層自在なものとなる。探索的データ解析(EDA)の過程では、前処理やフィルタリング、可視化を繰り返しながらデータの本質に迫り、最適な解析手法を見つけ出す。この過程では、Rの柔軟性を活かしつつ、試行錯誤を続けて解析の最適化を図ることが重要である。
最後に、これらの操作を円滑に行うためには、オブジェクトの詳細な構造を確認するための関数、例えばstr()
などを用いることが効果的である。これにより、複雑なデータ構造を理解し、効率的にデータを操作できるようになる。
| データ構造 | 種別 | 用途 | データ構造を扱うパッケージ | サブセットの方法 | データの基本処理のための主要な関数 | 適用可能な解析関数 |
|---|---|---|---|---|---|---|
| ベクトル(vector) | CRAN | 単一データ型の要素の集まり、基本的な統計計算やフィルタリング。 | base |
- [ や [[]] でのインデックス指定- 論理ベクトルでの条件指定 |
- 基本関数: mean(), sum(),
length(), sort(), unique()- 演算関数: +, -, *,
/, ^ |
- 統計関数: cor(), sd(),
var(), t.test() |
| リスト(list) | CRAN | 異なるデータ型や構造をまとめ、複雑なデータの管理に使用。 | base, purrr |
- [[ ]] でのインデックス指定- $
での名前指定 |
- リスト操作: lapply(), sapply()
(base)- リスト操作: map(),
map2(), pmap() (purrr) |
- リストの要素に関数を適用: lapply(),
map() |
| 行列(matrix) | CRAN | 数値データの統計解析や行列演算。 | base, Matrix |
- [ , ] でのインデックス指定- row() や
col() で行・列の抽出 |
- 行列演算: matrix(), %*%,
t(), solve()- 統計処理: prcomp() (stats) |
- 主成分分析: prcomp() (stats)- 共分散行列: cov() |
| データフレーム(data.frame) | CRAN | 異なるデータ型を持つ列を行列形式で管理し、データ解析の基本。 | base, dplyr, tidyr |
- - |
- データ操作: - データ整形: |
- 統計解析: - データ整形: |
| ティブル(tibble) | CRAN | データフレームの改良版、tidyverse
と連携しデータ解析に使用。 |
tibble, dplyr, tidyverse |
- - |
- データ操作: - ティブル変換: |
- データ整形: - データ解析: |
| igraph | CRAN | グラフデータ構造(ネットワーク)の管理と解析。 | igraph |
- グラフのサブセット: induced_subgraph(),
delete_vertices() |
- ノード操作: - エッジ操作: |
- ネットワーク解析: betweenness(),
closeness(), page_rank()
(igraph) |
| dgeMatrix(ディジーマトリックス) | CRAN | スパース行列の管理、行列演算や大規模データの解析に使用。 | Matrix, BiocParallel |
- - 行・列の名前指定: |
- 行列演算: - スパース行列の作成: |
- 大規模行列の解析: - 主成分分析: |
| SummarizedExperiment | Bioconductor | 遺伝子発現データやアノテーション情報の統合管理。 | SummarizedExperiment, DESeq2 |
- assay(), rowData(),
colData() を用いた抽出 |
- データ操作: - フィルタリング: |
- 発現変動解析: - データ変換: |
| ExpressionSet | Bioconductor | マイクロアレイやRNA-seqの発現データの統合管理。 | Biobase, limma |
- exprs(), pData(), fData()
を用いた抽出 |
- データ操作: - データ整形: |
- 発現変動解析: - 発現変動解析: |
| GRanges(GenomicRanges) | Bioconductor | 染色体位置情報の管理、ゲノムアノテーションやピークデータの操作。 | GenomicRanges, rtracklayer |
- GRanges() オブジェクトのメタ情報を用いた抽出 |
- ゲノム領域の操作: - 領域間の操作: |
- アノテーション解析: - 重複解析: |
| SingleCellExperiment | Bioconductor | シングルセルRNA-seqデータの統合管理。 | SingleCellExperiment, scran |
- counts(), colData(),
rowData() を用いた抽出 |
- データ操作: - 正規化: |
- 発現変動解析: - クラスタリング解析: |
| DGEList | Bioconductor | RNA-seqカウントデータの管理、発現変動解析の準備。 |
edgeR | - counts(), samples(),
genes() を用いた抽出 | - データ操作:
calcNormFactors(), estimateCommonDisp(),
estimateTagwiseDisp() (edgeR)- フィルタリング: filterByExpr() | - 発現変動解析:
exactTest(), glmFit(), glmLRT()
(edgeR)- 正規化: calcNormFactors()
(edgeR) | |
||||
| MSnSet(Mass Spectrometry Set) | Bioconductor | 質量分析データ(プロテオミクスデータ)の統合管理。 | MSnbase, MSstats |
- exprs(), pData(), fData()
を用いた抽出 |
- データ操作: exprs(), pData(),
fData() (MSnbase)- データ整形: normalize(), logTransform() |
- 発現変動解析: msqrob(), diffExp()
(MSstats)- 正規化: normalize()
(MSnbase) | |
| xcmsSet | Bioconductor | 質量分析データのピーク検出とアライメント。 | xcms, CAMERA |
- peaks(), group() を用いた抽出 |
- ピーク検出: findPeaks.centWave(),
findPeaks.massifquant() (xcms)- データ整形: group(), fillPeaks()
(xcms) |
-発現変動解析: diffreport() (CAMERA)- ピークアノテーション: annotate() (CAMERA) |
データ構造の理解が高度な解析の基礎
各解析において取り扱うデータの複雑性に応じて、非常に多様なデータ構造が定義されていることが理解できる。実際のところ、上記以外にもパッケージ特有のデータ構造が定義されている場合も多い。
したがって、あるパッケージの関数を基に解析フローを構築する場合、各関数の入力として適切なデータ構造に合わせたり、場合によっては自らデータ構造を構築したり、別の構造から変換する操作を行わなければならない。
また、出力結果をさらに別のパッケージの関数に入力する際、出力データから必要な情報を抽出し、データ構造を適切に調べ、操作して利用する能力が不可欠となる。この意味で、高度なデータ解析における基礎とは、データ構造を的確に理解し、自在に操作できるスキルであるといえる。
データ構造の理解と操作が解析の基盤となることを、具体的な例を用いてRにおけるデータ操作の本質を探っていく。ここで学ぶ操作は、複雑なデータ操作に対する基礎となることを強調しておく。
基本的なデータ構造と操作
Rにおける基本的なデータ構造は、ベクトル、行列、データフレーム、リストである。以下では、それらを順に説明する。
ベクトル(Vector)
Rの最も基本的なデータ構造であり、数値、文字列、論理値などを1次元で格納する。
c()関数を用いて作成し、要素の追加、削除、並べ替えなどの操作が可能である。例:
vec <- c(1, 2, 3, 4, 5) vec[3] # 3番目の要素を取得 vec[vec > 2] # 2より大きい要素を抽出行列(Matrix)
同じデータ型の要素を2次元で格納するデータ構造。
matrix()関数を用いて作成し、行列演算や部分行列の取得が可能である。例:
mat <- matrix(1:9, nrow = 3) mat[2, 3] # 2行3列目の要素を取得データフレーム(Data Frame)
異なるデータ型の列を持つことができる2次元データ構造。
data.frame()関数で作成し、列名や行名でアクセス可能である。表形式のデータを扱うのに最適な構造である。例:
df <- data.frame(name = c("Alice", "Bob", "Charlie"), age = c(25, 30, 35), score = c(80, 90, 85)) df$name # "name" 列を取得リスト(List)
異なるデータ型や構造を持つ要素を格納できるデータ構造。
list()関数を用いて作成し、リスト内の要素には名前やインデックスでアクセスできる。例:
lst <- list(name = "Alice", age = 25, scores = c(80, 90, 85)) lst$scores # ベクトル "scores" を取得
データ構造を理解し自在に操作する
これらのデータ構造を理解しただけでは、その真価は発揮されない。これらの構造は相補的なものであり、相互に連携して使用されることが多い。例えば、行列は数値データを効率的に格納するが、行列の操作にはベクトルが頻繁に用いられる。以下の例でその関係性を示す。
行列とベクトルの関係
行列とベクトルは、Rにおけるデータ操作の基本単位であり、これらを組み合わせることで効率的なデータ処理が可能となる。まず、行列とベクトルの操作をいくつかの例で見ていく。
例1: 行列の作成とベクトルでの列抽出
次に、2行3列の行列を作成し、ベクトルを用いて特定の列を抽出する操作を行う。
# 行列の作成(2行3列の行列)
matrix_data <- matrix(c(1, 2, 3, 4, 5, 6), nrow = 2, ncol = 3)
# 行列の表示
print(matrix_data) [,1] [,2] [,3]
[1,] 1 3 5
[2,] 2 4 6ここで、行列 matrix_data
が作成された。この行列の構造は、2行3列で、それぞれの要素が縦方向に1から6までの整数で構成されている。次に、ベクトルを使って特定の列を抽出してみよう。例えば、この行列
matrix_data の2列目と3列目を抽出する。
# 列を指定するベクトル
columns_to_extract <- c(2, 3)
# ベクトルを使って行列から特定の列を抽出
extracted_columns <- matrix_data[, columns_to_extract]
# 抽出した列の表示
print(extracted_columns) [,1] [,2]
[1,] 3 5
[2,] 4 6この例では、columns_to_extract というベクトルで抽出する列番号(2列目と3列目)を指定し、matrix_data から対応する列を取り出した。これにより、行列から特定の部分だけを効率的に操作できる。
例2: 行列の行ごとの和の計算
次に、行列の行ごとの和をベクトルを使って計算してみよう。例えば、各行の要素の合計を求める。
# 行ごとの和を計算
row_sums <- rowSums(matrix_data)
# 行ごとの和の表示
print(row_sums)[1] 9 12この例では、rowSums() 関数を使って、行列 matrix_data の各行の要素を合計し、新しいベクトル row_sums に結果を格納している。行ごとの和が、1行目は 9、2行目は 12 であることがわかる。
例3: 行列の要素にベクトルを用いた条件付き処理
行列の各要素に対して、条件に基づいた操作を行うことも可能である。例えば、行列の要素が3以上のものに対して、1を加算する操作を行う。
# 元の行列を表示
print(matrix_data) [,1] [,2] [,3]
[1,] 1 3 5
[2,] 2 4 6# 条件を満たす要素に1を加算
matrix_data[matrix_data >= 3] <- matrix_data[matrix_data >= 3] + 1
# 結果を表示
print(matrix_data) [,1] [,2] [,3]
[1,] 1 4 6
[2,] 2 5 7この例では、行列 matrix_data の中から、matrix_data >= 3 という条件を満たす要素(3以上の要素)を選択し、それらの要素に対して1を加算している。結果として、行列の該当要素がすべて1ずつ増加した。
これらの例からもわかるように、行列とベクトルを組み合わせることで、効率的にデータを操作できる。この基本的な操作は、より高度な解析の基盤となる。
行列の構造を操作するということは、ベクトルで操作するということ
先の例1では、行列の添字をベクトルとして扱うことにより、柔軟な行と列の指定が可能であることが直感的に理解できる。一方、例2と例3は、例1とは異なる観点からの例題である。この意味を深く理解することで、行列構造を操作するための本質的な理解が得られる。
行列は、行と列で構成された二次元の表形式データであるが、実際にはこの行列も内部的には「ベクトル」として保存されている。具体的には、行列は「列ベクトル」を縦に並べた構造を持ち、要素は一列ずつ順番にベクトルに格納されている。この仕組みを理解することで、行列の操作が実際にはベクトルを用いた操作の延長であることがわかる。
行列とベクトルの関係
Rでは、行列は単一のベクトルとして内部で管理されており、行列の要素は列方向に順番に並べられたベクトルである。この特性を理解すれば、行列操作の多くはベクトル操作の延長であり、行列の各要素に対する操作はベクトルとしての要素への操作として考えることができる。
行列の構成とベクトル操作の関係
例えば、以下のような行列 matrix_data を考える。
# 行列の作成(2行3列の行列)
matrix_data <- matrix(c(1, 2, 3, 4, 5, 6), nrow = 2, ncol = 3)
print(matrix_data) [,1] [,2] [,3]
[1,] 1 3 5
[2,] 2 4 6この行列 matrix_data
は、内部的には次のようなベクトルとして格納されている。
c(1, 2, 3, 4, 5, 6)このベクトルは、行列の各列を縦に並べた順序で格納されており、matrix()
関数によって2行3列の行列として整形されている。この行列の要素を操作する際には、Rは内部でこのベクトルに対して操作を行い、その結果を再び行列として表示する。
条件付き操作におけるベクトルの役割
行列の条件付き操作も、内部のベクトルに対する操作と考えることができる。先ほどの例では、行列の各要素に対して「値が3以上」であるかを確認し、その条件を満たす要素に対して1を加算した。この条件判定も内部的にはベクトルとして処理されており、具体的には次のような論理ベクトルを用いている。
# 条件に基づく論理ベクトルの生成
logical_vector <- matrix_data >= 3
print(logical_vector) [,1] [,2] [,3]
[1,] FALSE TRUE TRUE
[2,] FALSE TRUE TRUEこの論理ベクトルは、matrix_data
の各要素が3以上かどうかを判定したもので、行列の形をしたTRUE/FALSEのベクトルである。この論理ベクトルを使い、条件を満たす要素を選択し、そこに対して加算を行っている。
# 条件を満たす要素に1を加算
matrix_data[logical_vector] <- matrix_data[logical_vector] + 1
print(matrix_data) [,1] [,2] [,3]
[1,] 1 4 6
[2,] 2 5 7まとめると、
行列はベクトルの集合体: 行列は、内部的には一列に並べたベクトルとして保存されているため、行列に対する操作はベクトルに対する操作と同等に扱うことができる。
条件付き処理もベクトルとして処理: 行列の要素に条件を適用する際、内部では論理ベクトルが生成され、それを使って条件を満たす要素を選択して操作を行う。
ベクトル操作の延長としての行列操作: 行列操作は、ベクトルの要素に対する操作の延長として考えると理解しやすく、これは条件付き操作や行列の各行・各列を取り出して操作する場合にも適用できる。
このように、行列とベクトルの関係を理解することで、行列操作の背後にあるベクトル操作の考え方を把握し、より柔軟で効率的なデータ操作が可能になる。
他の構造に対する操作もベクトルによって操作する
他のデータ構造の操作においても、行列構造と同様に、ベクトルによる操作の延長であると考えられる。例えば、データフレームは、行列のような表形式であるが、行列と異なる点は、列ごとに異なるデータ型(数値型、文字列型などのRにおける情報の内部表現カテゴリ)を格納できるという点である。
データフレームにおけるベクトル操作の応用
列の選択やフィルタリング
データフレームの各列は内部的にはベクトルとして格納されており、行列と同様に、ベクトル操作を通じて効率的なデータ操作が可能である。例えば、特定の列だけを抽出したり、条件に基づいて行をフィルタリングする操作もベクトルを用いて行われる。
例1: 列の抽出
データフレームの特定の列(ベクトル)を抽出する例である。
# データフレームの作成
df <- data.frame(
ID = 1:5,
Name = c("Alice", "Bob", "Charlie", "David", "Eve"),
Score = c(85, 92, 78, 90, 87)
)
print(df) ID Name Score
1 1 Alice 85
2 2 Bob 92
3 3 Charlie 78
4 4 David 90
5 5 Eve 87# Name 列(ベクトル)を抽出
name_vector <- df[,c(2,3)]
print(name_vector) Name Score
1 Alice 85
2 Bob 92
3 Charlie 78
4 David 90
5 Eve 87この例では、データフレーム df の Name
列を抽出し、ベクトル name_vector
として格納した。データフレーム内の列はベクトルであり、他のベクトルと同様に操作することができる。
例2: 条件に基づく行のフィルタリング
データフレームの行を、ベクトルを用いて条件付きでフィルタリングする例である。
# Score が 85 以上の行を抽出
filtered_df <- df[df$Score >= 85, ]
print(filtered_df) ID Name Score
1 1 Alice 85
2 2 Bob 92
4 4 David 90
5 5 Eve 87この例では、Score 列の値が 85
以上である行をフィルタリングしている。この操作も、df$Score >= 85
という条件ベクトル(TRUE/FALSE)を用いて、該当する行を抽出するものである。
リストにおけるベクトル操作の応用
リストは、ベクトルを拡張したような構造を持ち、異なるデータ型や構造をまとめて保持できる柔軟なデータ構造である。データフレームや行列を含めて格納することもできる。基本的な構造の中で最も柔軟な構造を有しており、さまざまなパッケージ関数の出力値の構造としても用いられている。ここでもリスト内の各要素は、ベクトルとして操作することができる。
例1: ベクトルによる要素の選択
# 数値ベクトルを格納したリストの作成
num_list <- list(a = 1:5, b = 6:10, c = 11:15)
print(num_list)$a
[1] 1 2 3 4 5
$b
[1] 6 7 8 9 10
$c
[1] 11 12 13 14 15# 選択する要素の名前をベクトルとして定義
select_vector <- c("a", "c")
# リストから選択した要素を抽出
selected_elements <- num_list[select_vector]
print(selected_elements)$a
[1] 1 2 3 4 5
$c
[1] 11 12 13 14 15この例では、select_vector というベクトルを用いて、リスト
num_list から a と c
の要素を抽出している。リストの各要素名をベクトルとして指定し、必要な部分だけを抽出することで、目的のデータを簡単に取得できる。
例2: リスト内の各要素に対して操作を行う
# 数値ベクトルを格納したリストの作成
num_list <- list(a = 1:5, b = 6:10, c = 11:15)
print(num_list)$a
[1] 1 2 3 4 5
$b
[1] 6 7 8 9 10
$c
[1] 11 12 13 14 15# リスト内の各ベクトルに対して 2 を加算
modified_list <- lapply(num_list, function(x) x + 2)
print(modified_list)$a
[1] 3 4 5 6 7
$b
[1] 8 9 10 11 12
$c
[1] 13 14 15 16 17この例では、lapply 関数を用いて、リスト
num_list 内の各ベクトルに対して +2
の操作を行っている。lapply
関数は、リストの各要素に関数を適用し、その結果をリストとして返す。
例3: 条件付きフィルタリング
# 数値ベクトルを格納したリストの作成
num_list <- list(a = 1:5, b = 6:10, c = 11:15)
print(num_list)$a
[1] 1 2 3 4 5
$b
[1] 6 7 8 9 10
$c
[1] 11 12 13 14 15# 各ベクトルから、条件を満たす要素のみを抽出
filtered_list <- lapply(num_list, function(x) x[x > 8])
print(filtered_list)$a
integer(0)
$b
[1] 9 10
$c
[1] 11 12 13 14 15この例では、lapply を用いて、リスト
num_list 内の各ベクトルに対して、条件 x > 8
を満たす要素のみを抽出している。a
には条件を満たす要素がないため空のベクトル、b には 9 と
10、c には 11 以上の要素が抽出されている。
シングルセルデータにおけるベクトル操作の応用
SingleCellExperiment
オブジェクトは、シングルセルRNA-seqデータを管理するためのデータ構造であり、各細胞や遺伝子に関するアノテーション情報や、低次元データなどを統合的に管理する。このオブジェクトに対しても、ベクトル操作が重要である。
シングルセルデータの条件付きフィルタリング
シングルセルデータの各サンプル(細胞)に対して、条件に基づいたフィルタリングやサブセットの抽出を行う際に、ベクトル操作が役立つ。
例1: 細胞数のフィルタリング
library(SingleCellExperiment)
# SingleCellExperiment オブジェクトの作成(ダミーデータ)
sce <- SingleCellExperiment(
assays = list(counts = matrix(rpois(100, lambda = 5), nrow = 10, ncol = 10)),
colData = DataFrame(CellType = rep(c("A", "B"), each = 5))
)
# 各細胞の総カウント数を計算
total_counts <- colSums(counts(sce))
# 総カウント数が 20 以上の細胞を選択
sce_filtered <- sce[, total_counts >= 20]
print(dim(sce_filtered))[1] 10 9この例では、SingleCellExperiment
オブジェクトの各細胞の総カウント数を計算し、総カウント数が 20
以上の細胞をフィルタリングした。total_counts >= 20
という条件ベクトルを用いて、細胞をサブセット化している。
以上のように、データ構造にかかわらず、ベクトル操作はデータのサブセット化や変換、条件付き処理など、様々な場面で非常に重要な役割を果たす。これらの操作を適切に理解し、活用することで、より高度なデータ解析や操作が可能となる。
ベクトル操作のまとめ
さまざまなデータ構造において、ベクトル操作は基本的かつ非常に重要な役割を果たす。ベクトルは、Rのあらゆるデータ構造の基礎を成すものであり、データのサブセット化、フィルタリング、演算など、様々な操作を行うための基礎となることのイメージがついてきたと思う。重複する部分はあるが、いくつかの主要な異なるデータ構造において、普遍的に使用されるベクトル操作をまとめた(完全ではない)。
1. ベクトル基本操作
ベクトルは、Rの最も基本的なデータ構造であり、数値、文字列、論理値などの一連のデータを扱うために使用されます。
- ベクトルの生成:
c(1, 2, 3, 4, 5)、1:5、seq(1, 5, by = 1)など - 要素のアクセス:
x[1](1番目の要素を取得) - 条件付き抽出:
x[x > 3](3より大きい要素を抽出) - 要素の追加:
c(x, 6)(ベクトルの末尾に要素6を追加) - 演算:
x + 1(ベクトルの各要素に1を加算)
2. データフレームにおけるベクトル操作
データフレームは、異なるデータ型のベクトルを列として持つ二次元データ構造です。各列はベクトルとして扱われます。
- 列の抽出:
df$columnまたはdf[["column"]]で特定の列をベクトルとして取得 - 行の抽出:
df[1, ](1行目の全データを取得) - 条件付きフィルタリング:
df[df$column > 10, ](column列の値が10より大きい行を抽出) - 列の演算:
df$new_column <- df$column1 + df$column2(column1とcolumn2の和を新しい列として追加)
3. 行列におけるベクトル操作
行列は、同じデータ型を持つ二次元データ構造であり、行と列のインデックスを持つ点でベクトルと異なります。
- 行列の要素に対する操作:
matrix_data[1, ](1行目をベクトルとして抽出) - 条件付き置換:
matrix_data[matrix_data > 5] <- 0(行列の各要素に対して5より大きい要素を0に置換) - 行列積の演算:
matrix_data %*% c(1, 2, 3)(行列とベクトルの積) - 行列の列をベクトルとして抽出:
matrix_data[, 2](2列目をベクトルとして抽出)
4. リストにおけるベクトル操作
リストは、異なる型や長さを持つデータを要素として格納することができるデータ構造です。リストの各要素は、ベクトルとして扱うことができます。
- リスト内のベクトルの抽出:
list_data[[1]](リストの1番目の要素を抽出) - ベクトルを使ったフィルタリング:
lapply(list_data, function(x) x[x > 3])(リスト内の各ベクトルから、3より大きい要素を抽出) - リスト内のベクトルの長さを取得:
sapply(list_data, length)(リスト内の各ベクトルの長さを取得) - リスト内のベクトルの演算:
lapply(list_data, function(x) x + 1)(リスト内の各ベクトルに1を加算)
5. SummarizedExperimentやSingleCellExperimentにおけるベクトル操作
SummarizedExperiment や
SingleCellExperiment
は、遺伝子発現データとメタ情報を統合的に管理するデータ構造です。
- 遺伝子やサンプルのフィルタリング:
se[rowData(se)$gene == "TP53", ](遺伝子TP53に対応する行を抽出) - 特定サンプルの抽出:
se[, colData(se)$condition == "treated"](conditionがtreatedのサンプルのみ抽出) - 発現データへのベクトル操作:
assay(se)["gene1", ] <- assay(se)["gene1", ] + 1(遺伝子gene1の発現値に1を加算) - アノテーション情報の操作:
rowData(se)$type <- "oncogene"(全遺伝子のtype列にoncogeneとして注釈を追加)
6. DGEListにおけるベクトル操作
DGEList
は、発現変動解析を行うためのデータ構造で、遺伝子発現データと実験条件を格納します。
- 条件ごとのデータ抽出:
dge$samples[group == "control", ](コントロール群のサンプル情報を抽出) - 発現データのフィルタリング:
dge$counts[dge$counts > 5](発現量が5を超える遺伝子のみを抽出) - サンプルの正規化:
dge$samples$norm.factors <- calcNormFactors(dge)(サンプル間の正規化係数を計算して格納)
シークエンサーの定量データを管理するためのオブジェクト構造
BioconductorのSummarizedExperimentやSingleCellExperimentといったデータ構造は、バイオインフォマティクス解析において非常に有用である。これらのオブジェクトは、定量値(発現データなど)、メタ情報(サンプルや遺伝子の情報)、アノテーション情報(遺伝子や領域の機能的説明)を一つのオブジェクト内で統一的に管理できるため、解析の整合性を確保しながらデータの操作を効率化できる。
これらのデータ構造の大きな利点は、例えばサンプルや遺伝子のフィルタリング(ベクトル操作によって!)を行った際に、すべての関連情報が自動的に更新される点である。これにより、複数のデータセット間でデータの一貫性を保つ手間が省け、解析作業が大幅に簡易化される。これらの特徴は、複雑なバイオインフォマティクスデータを扱う際に非常に重要である。下記に利点をまとめた。
データの整合性を保つ: Assayデータとメタデータ、アノテーションデータを一元管理することで、データの整合性を保ちやすくなる。これにより、解析におけるデータの混乱やエラーを防ぐことができる。
柔軟なデータ操作: データ構造に対する理解が深まると、解析フローの中で必要に応じてデータを自由に操作し、適切なタイミングでアノテーションを追加したり、サンプル情報を調整することが可能になる。
他の解析パッケージとの連携:
SummarizedExperimentは、他のバイオインフォマティクス解析パッケージ(例:DESeq2,edgeRなど)と連携が可能であり、解析パイプライン全体を効率化できる。
なお、これらのデータ構造の前身として ExperimentSet
という形式があった。現在では、SummarizedExperiment
などに多く置き換わっているが、依然として ExperimentSet
が使われている場面もある。例えば、公共データベースのGEO(Gene Expression
Omnibus)からデータを取得するためのGEOqueryパッケージでは、マイクロアレイデータを含む場合に
ExperimentSet
が使用されることがある。そのような場合、より扱いやすく、最新の解析パイプラインと互換性のある
SummarizedExperiment
に変換して利用することが推奨される。これにより、Bioconductorエコシステム内の他のパッケージと容易に統合でき、解析の柔軟性と効率性を高めることができる。
SummarizedExperiment

図1: SummarizedExperimentオブジェクトの構造
1. SummarizedExperimentの基本的なデータ構造
SummarizedExperiment
オブジェクトは、以下の3つの主要なコンポーネントから構成されている。
Assays: 実際のデータ(遺伝子発現量やタンパク質量など)が含まれている行列である。行が遺伝子(またはその他の特徴量)に対応し、列がサンプルに対応する。
Row Data: 各行(遺伝子)に対応するアノテーション情報(遺伝子名や機能、アノテーションなど)を格納する。
Column Data: 各列(サンプル)に対応するメタデータ(サンプルの状態や実験条件、性別、年齢など)を格納する。
このデータ構造を活用することで、遺伝子発現量と、それに関連するサンプル情報やアノテーションを一体として管理・解析することができる。
SummarizedExperimentオブジェクトの作成
まず、データをもとに SummarizedExperiment
オブジェクトを作成する基本的な操作を示す。
# SummarizedExperiment パッケージをロード
library(SummarizedExperiment)
# 発現データ (行が遺伝子、列がサンプル)
assay_data <- matrix(rnorm(1000), nrow=100, ncol=10)
# サンプルに関するメタデータ
col_data <- DataFrame(
condition = rep(c("Control", "Treatment"), each=5),
age = c(30, 32, 29, 45, 50, 28, 36, 33, 44, 40)
)
# 遺伝子に関するアノテーションデータ
row_data <- DataFrame(
gene_name = paste0("Gene", 1:100),
chromosome = rep(c("chr1", "chr2", "chr3"), length.out=100)
)
# SummarizedExperiment オブジェクトを作成
se <- SummarizedExperiment(assays = list(counts = assay_data),
rowData = row_data,
colData = col_data)
# 結果の確認
seこの操作で、遺伝子発現データと関連するメタデータを1つのSummarizedExperimentオブジェクトにまとめることができる。
2. SummarizedExperimentの基本的な操作
Assayデータの操作
Assayデータは、遺伝子発現量などの数値データが含まれる行列であり、各遺伝子(行)と各サンプル(列)に対応する。
- Assayデータの取得
# Assayデータを取得
assay_data <- assay(se)
head(assay_data)- Assayデータにアクセスして変更
# Assayデータの変更
assay(se)[1, ] <- rnorm(10)Row Dataの操作
Row Data は、各遺伝子に関連するアノテーション情報(例えば、遺伝子名や染色体情報)が格納されている部分である。
- Row Dataの取得
# Row Dataの取得
row_data <- rowData(se)
head(row_data)- Row Dataの変更
# 遺伝子アノテーションの変更
rowData(se)$gene_name[1] <- "NewGene1"Column Dataの操作
Column Data は、各サンプルに関する情報(例えば、サンプルの状態、性別、年齢など)が含まれている。
- Column Dataの取得
# Column Dataの取得
col_data <- colData(se)
head(col_data)- Column Dataの変更
# サンプルメタデータの変更
colData(se)$condition[1] <- "NewCondition"4. エラー処理と問題解決
データを扱う際に、期待した形式でデータが取得できない場合もある。その場合は、データの型や構造を確認し、問題を特定するスキルが必要である。以下にエラー処理の基本的な考え方を示す。
- データ構造が正しいかを確認する:
# データ構造を確認
str(se)####str() 関数の結果を読み解く
また、str()
関数は、データ構造の概要を簡潔に表示するための強力なツールでもある。特に複雑なデータ構造を持つ
SummarizedExperiment や SingleCellExperiment
のようなオブジェクトをはじめて扱う場合、その内部構造を理解することは、解析を進める上で不可欠である。ここでは、str()
関数の結果をどのように読み解くか、具体的な例を挙げて説明する。str()
関数を用いると、以下の情報が表示される
- データのクラス:
オブジェクトがどのクラスに属しているかを示す。例えば、
SummarizedExperimentやSingleCellExperimentといった特定のクラスである。 - オブジェクトの長さと構造: 各要素の数や階層構造が表示される。データフレームであれば、行数と列数、リストであれば各要素の個数などがわかる。
- 各要素の型と内容: 各要素がどのデータ型(数値、文字列、因子、リストなど)で、どのようなデータを含んでいるかが示される。
SummarizedExperiment オブジェクトの str()
結果の読み解き方
SummarizedExperiment
オブジェクトは、発現データ(アッセイデータ)と、遺伝子情報(rowData)、サンプル情報(colData)を統合的に管理するための構造である。以下は、SummarizedExperiment
オブジェクトの str() の出力例である。
library(SummarizedExperiment)
# サンプルデータを作成
counts <- matrix(rpois(100, lambda = 10), ncol = 5)
rownames(counts) <- paste0("gene", 1:20)
colData <- DataFrame(condition = c("A", "A", "B", "B", "C"))
se <- SummarizedExperiment(assays = list(counts = counts), colData = colData)
# str() でオブジェクトの構造を表示
str(se)出力例の解釈
Formal class 'SummarizedExperiment' [package "SummarizedExperiment"] with 5 slots
..@ assays :Formal class 'Assays' [package "SummarizedExperiment"] with 1 slot
.. ..@ data:List of 1
.. .. ..$ counts: int [1:20, 1:5] 15 8 7 14 12 10 11 7 16 12 ...
..@ rowData :Formal class 'DFrame' [package "S4Vectors"] with 6 slots
..@ colData :Formal class 'DataFrame' [package "S4Vectors"] with 6 slots
.. .. ..$ condition: Factor w/ 3 levels "A","B","C": 1 1 2 2 3
..@ metadata :list()
..@ NAMES :NULL
..@ elementMetadata: NULL- Formal class ‘SummarizedExperiment’:
このオブジェクトは、
SummarizedExperimentクラスであることを示している。 - @ assays:
発現データを格納するスロット。
list型のdataには、countsという名前の行列が含まれており、20行5列(20個の遺伝子、5つのサンプル)の整数値(int型)が入っています。発現値の例として、15, 8, 7, 14, 12, ...と表示されている。 - @ rowData: 各遺伝子に関する情報を格納するスロットです。今回は特に情報が格納されていないので詳細は表示されていませんが、通常は遺伝子名やアノテーション情報が含まれている。
- @ colData:
サンプルに関する情報を格納するスロットです。ここでは
conditionという因子(Factor)変数があり、3つのレベル(“A”, “B”, “C”)を持つことがわかる。 - @ metadata: メタデータを格納するリストで、今回は空である。
- @ NAMES: 要素の名前を格納するスロットで、今回は
NULLである。 - @ elementMetadata:
要素メタデータを格納するスロットで、今回は
NULLである。
以下の内容を「だ」「である」調で修正する:
SummarizedExperimentデータ変更の方法について
SummarizedExperiment
オブジェクトにおいてデータの変更を行う際には、assays、rowData、colData
といったスロットに格納されているデータを操作することが基本である。これらのスロットに格納されたデータは、通常のRのデータフレームや行列と同様に扱うことができ、適切な関数を使ってデータの追加や変更が可能である。以下に、各スロットに対するデータの変更方法を具体例とともに説明する。
1. Assayデータの変更
assays
スロットには、遺伝子発現データなどの数値データ(行列)が格納されている。行ごと(遺伝子)や列ごと(サンプル)のデータを直接変更したり、新たなアッセイデータを追加することが可能である。
Assayデータの取得と変更
# Assayデータの取得
assay_data <- assay(se)
# 例: 最初の遺伝子の発現データを全てゼロに変更
assay(se)[1, ] <- 0新たなAssayデータの追加
# 正規化された新たなアッセイデータを追加
normalized_counts <- log2(assay_data + 1)
assays(se)$normalized_counts <- normalized_countsこのように、元のカウントデータ(counts)に加えて、正規化されたデータ(normalized_counts)を別のアッセイとして追加することができる。
2. Row Dataの変更
rowData
スロットには、各遺伝子に関連するアノテーション情報が格納されている。例えば、遺伝子名や遺伝子機能の情報を追加・変更することができる。
Row Dataの取得と変更
# Row Dataの取得
row_data <- rowData(se)
# 例: 最初の遺伝子名を "NewGene1" に変更
rowData(se)$gene_name[1] <- "NewGene1"新たなRow Dataの追加
# 新しいアノテーションデータ(例: パスウェイ情報)を追加
rowData(se)$pathway <- c("Pathway1", "Pathway2", "Pathway3", ..., "Pathway100")3. Column Dataの変更
colData
スロットには、各サンプル(列)に関するメタ情報が格納されている。サンプルの状態や実験条件などを変更したり、追加することができる。
Column Dataの取得と変更
# Column Dataの取得
col_data <- colData(se)
# 例: 最初のサンプルの条件を "NewCondition" に変更
colData(se)$condition[1] <- "NewCondition"新たなColumn Dataの追加
# 新しいメタデータ(例: サンプルの処理時間)を追加
colData(se)$treatment_time <- c(24, 48, 72, 24, 48)4. データの削除
特定の行や列を削除することで、不要な遺伝子やサンプルを取り除くことも可能である。SummarizedExperiment
オブジェクトは柔軟に行や列を削除できる。
特定の遺伝子やサンプルの削除
# 例: 最初の遺伝子を削除
se <- se[-1, ]
# 例: 最初のサンプルを削除
se <- se[, -1]5. データの確認とエラー処理
データを変更した後は、str() や head()
などの関数を使ってデータ構造が正しく変更されたかを確認する必要がある。エラーが発生した場合、データの型や構造を確認し、問題を特定するスキルが求められる。
# データ構造の確認
str(se)このようにSummarizedExperiment
オブジェクトは、発現データとアノテーション情報、メタデータを統合的に管理できる便利なデータ構造である。データを追加・変更・削除する操作を習得することは、バイオインフォマティクス解析における重要なスキルであり、複雑なデータセットを扱う際に不可欠である。
SummarizedExperiment
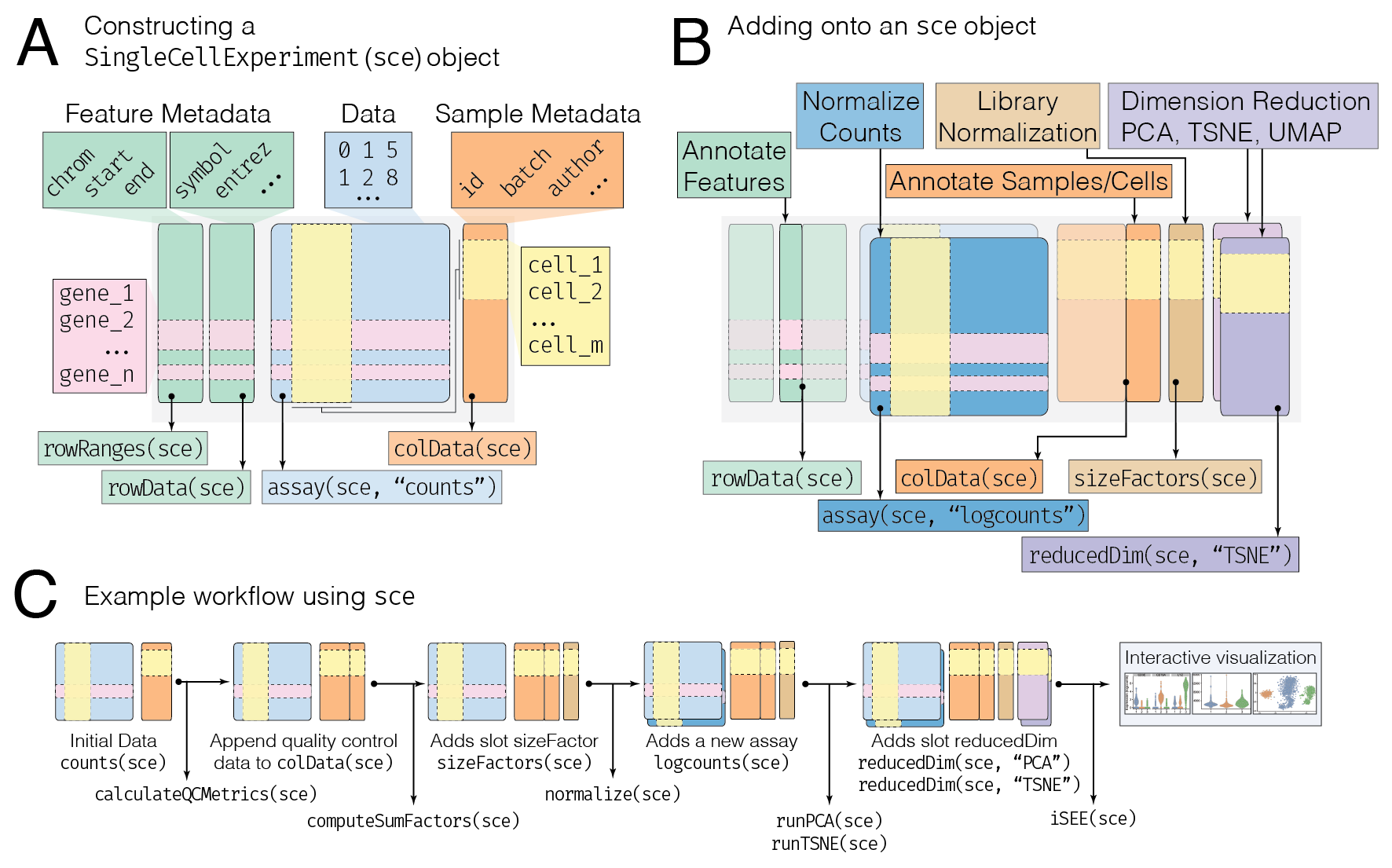
図2: SingleCellExperimentオブジェクトの構造
SingleCellExperiment
オブジェクトはSummarizedExperimentの基盤のうえに作られたもので、シングルセルRNAシーケンスデータを管理・解析するために設計されたデータ構造で、SummarizedExperimentと同様に以下の3つの主要なコンポーネントを持っている。
- Assays: 遺伝子発現量などのデータが含まれる行列。行が遺伝子や特徴量に対応し、列が細胞に対応する。
- Row Data: 各遺伝子に関連するアノテーション情報(遺伝子名や機能、アノテーションなど)を格納する。
- Column Data: 各細胞に対応するメタデータ(細胞の状態やクラスター情報など)を格納する。
これらの構造を活用することで、細胞ごとの遺伝子発現量と、サンプルや細胞に関連するメタデータを一体として管理し、解析することが可能である。また、次元削減の解析を行なうと、これに加えてreducedDimsのスロットが追加さたり、拡張要素が存在する。
SingleCellExperimentオブジェクトの作成
まず、シングルセルRNAシーケンスデータを基に
SingleCellExperiment オブジェクトを作成する方法を示す。
# 必要なパッケージをロード
library(SingleCellExperiment)
# 発現データ (行が遺伝子、列が細胞)
assay_data <- matrix(rnorm(1000), nrow=100, ncol=10)
# 細胞に関するメタデータ
col_data <- DataFrame(
cluster = rep(c("Cluster1", "Cluster2"), each=5),
condition = rep(c("Control", "Treatment"), each=5)
)
# 遺伝子に関するアノテーションデータ
row_data <- DataFrame(
gene_name = paste0("Gene", 1:100),
chromosome = rep(c("chr1", "chr2", "chr3"), length.out=100)
)
# SingleCellExperiment オブジェクトを作成
sce <- SingleCellExperiment(assays = list(counts = assay_data),
rowData = row_data,
colData = col_data)
# 結果の確認
sceこの操作により、遺伝子発現データや関連するメタデータを
SingleCellExperiment
オブジェクトとしてまとめ、シングルセルデータを統合的に管理できる。
2. SingleCellExperiment の基本的な操作
操作自体は、SummarizedExperimentとさほど違いはないが、下記にまとめる。
Assay データの操作
Assay データ には遺伝子発現量や特徴量データが格納されており、行が遺伝子、列が細胞に対応している。
- Assay データの取得
# Assay データを取得
assay_data <- assay(sce)
head(assay_data)- Assay データにアクセスして変更
# Assay データの変更
assay(sce)[1, ] <- rnorm(10)Row Data の操作
Row Data は、各遺伝子に関連するアノテーション情報(例: 遺伝子名、染色体など)が格納されている部分である。
- Row Data の取得
# Row Data の取得
row_data <- rowData(sce)
head(row_data)- Row Data の変更
# 遺伝子アノテーションの変更
rowData(sce)$gene_name[1] <- "NewGene1"Column Data の操作
Column Data は、各細胞に関するメタデータ(例: クラスター情報、サンプル条件など)が格納されている。
- Column Data の取得
# Column Data の取得
col_data <- colData(sce)
head(col_data)- Column Data の変更
# 細胞メタデータの変更
colData(sce)$condition[1] <- "NewCondition"3. str() 関数を用いたデータ構造の確認
SingleCellExperiment
オブジェクトの複雑なデータ構造を理解するには、str()
関数を使って内部構造を確認する。
# オブジェクトのデータ構造を確認
str(sce)出力例の解釈
Formal class 'SingleCellExperiment' [package "SingleCellExperiment"] with 6 slots
..@ int_elementMetadata: Formal class 'DataFrame' [package "S4Vectors"] with 6 slots
..@ colData : Formal class 'DataFrame' [package "S4Vectors"] with 6 slots
..@ reducedDims : Formal class 'SimpleList' [package "S4Vectors"] with 1 slot
..@ assays :Formal class 'Assays' [package "SummarizedExperiment"] with 1 slot
.. ..@ data:List of 1
.. .. ..$ counts: int [1:10, 1:10] 14 10 15 14 5 12 16 16 5 7 ...
..@ metadata : list()
..@ int_colData : Formal class 'DataFrame' [package "S4Vectors"] with 6 slots- Formal class ‘SingleCellExperiment’:
このオブジェクトは、
SingleCellExperimentクラスであることを示しています。 - @ int_elementMetadata: 内部的な遺伝子情報を格納するスロット。特に重要な情報は含まれていない場合があります。
- @ colData: サンプル情報(例えば、細胞情報)を格納するスロットです。デフォルトでは空ですが、通常は細胞のクラスター情報やメタデータが入ります。
- @ reducedDims:
次元削減データ(例えば、PCAやt-SNEの結果)を格納するスロットです。
SimpleList形式で管理され、各次元削減手法の結果がリストに格納されます。 - @ assays: 発現データを格納するスロット。ここには
countsという行列(10行10列)が含まれています。 - @ metadata: メタデータを格納するリストで、今回は空です。
- @ int_colData: 内部的なサンプルメタデータを格納するスロットです。
SingleCellExperimentのデータ変更方法について
SingleCellExperiment
オブジェクトも、SummarizedExperiment
に基づいたデータ構造を持ち、基本的なデータ操作方法は
SummarizedExperiment
と共通している。しかし、SingleCellExperiment
では、特にシングルセルデータに特化したスロットが追加されているため、その扱いについても説明する。ここでは、SingleCellExperiment
における各スロットのデータ変更方法を具体例を用いて説明する。
1. Assayデータの変更
SingleCellExperiment オブジェクトの assays
スロットには、各遺伝子と各サンプルの発現データ(行列)が格納されている。遺伝子ごとのデータを変更したり、新しいアッセイデータを追加することができる。
Assayデータの取得と変更
# Assayデータの取得
assay_data <- assay(sce)
# 例: 最初の遺伝子の発現データを全てゼロに変更
assay(sce)[1, ] <- 0新たなAssayデータの追加
# 正規化された新たなアッセイデータを追加
normalized_counts <- log2(assay_data + 1)
assays(sce)$normalized_counts <- normalized_countsこのように、新たなアッセイデータを簡単に追加することができ、元のデータを残しつつ異なる処理結果を保存することができる。
2. Row Dataの変更
rowData
スロットには、各遺伝子に対応するアノテーションデータが格納されている。ここでは遺伝子名や染色体情報などを操作することができる。
Row Dataの取得と変更
# Row Dataの取得
row_data <- rowData(sce)
# 例: 最初の遺伝子名を "NewGene1" に変更
rowData(sce)$gene_name[1] <- "NewGene1"新たなRow Dataの追加
# 新しいアノテーションデータを追加(例: パスウェイ情報)
rowData(sce)$pathway <- c("Pathway1", "Pathway2", "Pathway3", ..., "Pathway100")この操作で、各遺伝子に新たな注釈情報を追加することができる。
3. Column Dataの変更
colData
スロットには、各サンプルに対応するメタデータが格納されている。例えば、サンプルの処理条件や実験条件をここで管理する。
Column Dataの取得と変更
# Column Dataの取得
col_data <- colData(sce)
# 例: 最初のサンプルの条件を "NewCondition" に変更
colData(sce)$condition[1] <- "NewCondition"新たなColumn Dataの追加
# 新しいメタデータ(例: サンプルの処理時間)を追加
colData(sce)$treatment_time <- c(24, 48, 72, 24, 48)このようにして、各サンプルに関連する追加情報を格納することができる。
4. Reduced Dimensionの操作
SingleCellExperiment では、次元削減結果を
reducedDims
スロットに格納することができる。次元削減結果は、UMAPやPCAなどの結果を保存し、後からアクセス可能にする。
次元削減データの追加と取得
# PCA結果を追加
pca_result <- prcomp(t(assay(sce)))
reducedDims(sce)$PCA <- pca_result$x
# UMAP結果を取得
umap_result <- reducedDims(sce)$UMAP次元削減結果は簡単に追加・取得でき、可視化やクラスタリング解析に利用できる。
5. データの削除
SingleCellExperiment
オブジェクトにおいても、不要な行(遺伝子)や列(サンプル)を削除することが可能である。
特定の遺伝子やサンプルの削除
# 例: 最初の遺伝子を削除
sce <- sce[-1, ]
# 例: 最初のサンプルを削除
sce <- sce[, -1]この操作により、不要な遺伝子やサンプルを取り除き、データセットを整えることができる。
6. データの確認とエラー処理
データを変更した後は、データ構造が期待通りになっているかを確認する必要がある。特に次元削減やメタデータの変更は、解析に大きな影響を与えるため、str()
や summary() 関数を活用して確認する。
# データ構造の確認
str(sce)このようにSingleCellExperiment
オブジェクトは、シングルセルデータを効率的に管理・操作するための強力なデータ構造である。データの追加や変更、削除を適切に行うことで、複雑な解析をスムーズに進めることができる。SummarizedExperiment
に比べて、次元削減結果やシングルセル特有のメタ情報を扱う機能が追加されているため、シングルセル解析には不可欠なツールとなっている。
データの結合
Rにおけるデータ操作を習熟するための2つ目の考え方は、異なるデータを結合する操作である。ここで結合という意味は、二つ以上のデータセット間で整合性を保ちながら、それらを一つに結びつけることを指す。データの結合は、データ解析やレポート作成において、異なる情報を統合して一貫性のある分析を行うために非常に重要である。結合の操作には、以下のような多様な方法がある。
1. 行方向の結合(縦結合)
行方向の結合は、同じ列構造を持つ二つ以上のデータフレームや行列を、行単位で連結する方法です。例えば、異なる実験条件や異なるサンプルのデータを一つのデータセットとして統合する際に用います。
関数の例
rbind():- 例:
rbind(df1, df2) - 同じ列名を持つ二つのデータフレームを、行方向に結合する。
- 例:
bind_rows()(dplyrパッケージ):- 例:
bind_rows(df1, df2) rbind()と似ているが、異なる列名を持つデータフレームでも結合でき、データフレーム同士の不一致を柔軟に扱います。
- 例:
使用例
# データフレームの作成
df1 <- data.frame(ID = 1:3, Name = c("Alice", "Bob", "Charlie"))
df2 <- data.frame(ID = 4:6, Name = c("David", "Eve", "Frank"))
# 行方向の結合
df_combined <- rbind(df1, df2)
print(df_combined) ID Name
1 1 Alice
2 2 Bob
3 3 Charlie
4 4 David
5 5 Eve
6 6 Frank2. 列方向の結合(横結合)
列方向の結合は、同じ行数を持つ二つ以上のデータフレームや行列を、列単位で結びつける方法です。これにより、同じサンプルに対して異なる特徴(変数)を追加することができます。
関数の例
cbind():- 例:
cbind(df1, df2) - 同じ行数を持つ二つのデータフレームや行列を列方向に結合します。
- 例:
bind_cols()(dplyrパッケージ):- 例:
bind_cols(df1, df2) - 列名が衝突する場合には自動的に解決し、柔軟に列結合が可能です。
- 例:
使用例
# データフレームの作成
df1 <- data.frame(ID = 1:3, Score = c(85, 90, 88))
df2 <- data.frame(Name = c("Alice", "Bob", "Charlie"), Age = c(25, 30, 28))
# 列方向の結合
df_combined <- cbind(df1, df2)
print(df_combined) ID Score Name Age
1 1 85 Alice 25
2 2 90 Bob 30
3 3 88 Charlie 283. キーによる結合(データベース結合)
異なるデータセット間の共通のキー(IDや名前など)を基に、関連するデータを結びつける方法である。SQLのJOINに類似しており、データ解析において非常に重要な操作である。特に、異なるデータソースから取得した情報を組み合わせて、新しいデータセットを作成する際に使用する。Rでは、merge()
関数や dplyr パッケージの left_join(),
inner_join(), full_join()
などの関数を使用して、さまざまな結合を行う。
関数の例
merge():- 例:
merge(df1, df2, by = "ID") - 共通の列(キー)を基に二つのデータフレームを結合する。内部結合、外部結合、左結合、右結合が可能である。
- 例:
left_join(),inner_join(),full_join()(dplyrパッケージ):- 例:
left_join(df1, df2, by = "ID") - 結合方法に応じて、左結合、内部結合、全結合など、柔軟に結合のタイプを指定できる。
- 例:
使用例
# データフレームの作成
df1 <- data.frame(ID = 1:3, Name = c("Alice", "Bob", "Charlie"))
df2 <- data.frame(ID = 2:4, Score = c(92, 85, 78))
# キーによる結合
df_combined <- merge(df1, df2, by = "ID")
print(df_combined) ID Name Score
1 2 Bob 92
2 3 Charlie 85結合タイプごとの説明
# サンプルデータの作成
df1 <- data.frame(
ID = c(1, 2, 3, 4),
Name = c("Alice", "Bob", "Charlie", "David"),
Score = c(85, 92, 78, 90)
)
df2 <- data.frame(
ID = c(2, 3, 5),
Subject = c("Math", "Science", "History"),
Grade = c("A", "B", "C")
)
1. **左結合(Left Join)**
左側のデータセット(`df1`)を基準として、右側のデータセット(`df2`)の対応するキーを持つ行を結合する。右側のデータに一致する行がない場合でも、左側のデータはすべて保持され、対応する右側のデータは`NA`(欠損値)として扱われる。
# 左結合 (left_join)
left_join(df1, df2, by = "ID") ID Name Score Subject Grade
1 1 Alice 85 <NA> <NA>
2 2 Bob 92 Math A
3 3 Charlie 78 Science B
4 4 David 90 <NA> <NA>この例では、df1 のすべての行が保持され、df2
に対応する行がある場合は、その情報が追加される。
- 内部結合(Inner Join)
両方のデータセット(df1
とdf2)で共通のキーを持つ行のみを結合する。どちらのデータにも存在する行のみを抽出し、それ以外の行は除外される。
# 内部結合 (inner_join)
inner_join(df1, df2, by = "ID") ID Name Score Subject Grade
1 2 Bob 92 Math A
2 3 Charlie 78 Science Bこの例では、df1 と df2
の両方に存在するキーの行のみが結合され、両方に存在しない行は除外される。df1
と df2 の ID
が共通している行(2と3)のみが表示されている。
- 全結合(Full Join)
両方のデータセットのすべての行を保持し、いずれか一方にしか存在しない場合は、
NAとしてデータを埋める。これにより、データセットの欠損部分を把握しやすくなる。
# 全結合 (full_join)
full_join(df1, df2, by = "ID") ID Name Score Subject Grade
1 1 Alice 85 <NA> <NA>
2 2 Bob 92 Math A
3 3 Charlie 78 Science B
4 4 David 90 <NA> <NA>
5 5 <NA> <NA> History Cdf1 と df2
のすべての行が保持され、欠損部分は NA
で埋められる。この例では、df1 と df2
のすべての行を含むデータセットが作成され、対応するデータがない場合は
NA として表示される。
- 右結合(Right Join)
右側のデータセット(df2)を基準として、左側のデータセット(df1)の対応するキーを持つ行を結合する。左側のデータに一致する行がない場合でも、右側のデータはすべて保持され、対応する左側のデータは
NA として扱われる。
``` r
library(dplyr)
right_join(df1, df2, by = "ID")
```この例では、df2 のすべての行が保持され、df1
に対応する行がある場合は、その情報が追加されている。
- アンチ結合(Anti Join)
左側のデータセット(df1)に存在し、右側のデータセット(df2)に存在しない行のみを抽出する。異なるデータの把握や欠損値の確認に有効である。
# アンチ結合 (anti_join)
anti_join(df1, df2, by = "ID") ID Name Score
1 1 Alice 85
2 4 David 90この例では、df1 に存在するが、df2
に存在しない行のみが抽出される。df1 に存在するが
df2 に存在しない ID
の行のみが表示されている(ID: 1 と 4)。
これらの結合操作は、異なるデータソースからの情報を組み合わせて新しい知見を得る際に非常に重要であり、データ解析の初期段階で頻繁に使用されている。各結合タイプを理解し、適切に選択することが、効率的で正確なデータ解析を行うための鍵となる。
4. 結合における注意点
データ結合の際には、以下の点に注意する必要がある。
- 共通キーの有無: データセット間で共通のキー(IDなど)が存在しない場合、正しい結合ができない。事前にデータを確認し、必要に応じてキーを統一する。
- 欠損値の扱い: 外部結合(left join, full join)では、共通のキーを持たない行に欠損値(NA)が発生する。これらの欠損値の扱いを事前に考慮しておくことが重要である。
- 列名の重複: 列名が重複する場合、自動的に名前が変更されるか、エラーが発生する。結合前に列名を確認し、適切に調整することが推奨される。
演習:SummarizedExperimentとSingleCellExperimentにおけるデータ結合操作
次の演習では、SummarizedExperiment と
SingleCellExperiment
オブジェクトを使ったデータ結合操作を学ぶ。この演習では、外部アノテーションデータを利用してオブジェクトに情報を統合する方法や、複数のシングルセルデータを結合する方法を扱う。
演習1: HGNCデータベースからのアノテーション追加(SummarizedExperiment)
- 以下のコードを実行して、
SummarizedExperimentオブジェクトを作成し、仮のHGNCデータを使用して遺伝子シンボルを追加する。 left_join()関数の役割を理解し、SummarizedExperimentのrowDataに遺伝子シンボルが正しく統合されているか確認する。
library(SummarizedExperiment)
# サンプルデータを作成
counts <- matrix(rpois(100, lambda = 10), ncol = 5)
rownames(counts) <- paste0("gene", 1:20) # 仮の遺伝子ID
colData <- DataFrame(condition = c("A", "A", "B", "B", "C"))
se <- SummarizedExperiment(assays = list(counts = counts), colData = colData)
# 外部アノテーションデータ
hgnc_data <- data.frame(
GeneID = paste0("gene", 1:20),
GeneSymbol = paste0("Symbol", 1:20),
stringsAsFactors = FALSE
)
# パイプラインを使わない方法で、遺伝子IDをキーにアノテーションを追加
row_data <- as.data.frame(rowData(se)) # rowDataをデータフレームに変換
joined_data <- left_join(row_data, hgnc_data, by = c("rowname" = "GeneID")) # left_joinを使って結合
rowData(se) <- DataFrame(joined_data) # 結果をSummarizedExperimentオブジェクトに反映
# パイプラインを使ったコードの例
# rowData(se) <- rowData(se) %>%
# as.data.frame() %>%
# left_join(hgnc_data, by = c("rowname" = "GeneID")) %>%
# DataFrame()
# merge() を使った結合の例
# row_data <- as.data.frame(rowData(se)) # rowDataをデータフレームに変換
# merged_data <- merge(row_data, hgnc_data, by.x = "row.names", by.y = "GeneID", all.x = TRUE) # merge() を使って結合
# rownames(merged_data) <- merged_data$Row.names # 行名を戻す
# merged_data$Row.names <- NULL # 一時的な列を削除
# rowData(se) <- DataFrame(merged_data) # 結果をSummarizedExperimentオブジェクトに反映
# 結果を表示
print(rowData(se))
確認事項:
left_join()を使用することで、オリジナルの遺伝子情報を保持しつつ、対応するアノテーションを追加できているか確認する。他の結合方法との比較を考察する
inner_join(): 両方のデータに共通するキーを持つ行のみを結合するため、アッセイデータからアノテーションのない遺伝子は除外されてしまう。このため、データの損失が発生しやすく、解析結果に影響を与える可能性がある。full_join(): 左右両方のデータセットのすべての行を保持するが、実際にはアノテーションデータ側にしかない情報はあまり使わないため、left_join()の方が適している場合が多い。right_join(): 右側のデータセット(外部アノテーションデータ)を基準にするが、バイオインフォマティクス解析においては、アッセイデータを基準に情報を追加することが一般的であるため、こちらも適用される場面は少ない。merge():merge()関数は、データフレームを結合するための標準的なRの関数であり、デフォルトではinner_join()と同じ動作(共通のキーを持つ行のみを残す)を行うが、all.x = TRUEを指定することでleft_join()と同様の動作を実現できる。キー名が異なる場合でも、by.xおよびby.y引数を用いることで対応できる。R標準の関数であり、使い慣れている場合には便利であるが、dplyrのleft_join()に比べてコードがやや冗長になることがある。
演習2: Ensemblデータベースからのアノテーション追加(SummarizedExperiment)
biomaRtパッケージを使用して、Ensemblから遺伝子シンボル情報を取得し、SummarizedExperimentオブジェクトに統合する。getBM()関数の使い方と、left_join()を使用してアノテーションを統合するプロセスを確認する。
library(SummarizedExperiment)
library(dplyr)
library(biomaRt)
# サンプルデータ作成
counts <- matrix(rpois(100, lambda = 10), ncol = 5)
rownames(counts) <- paste0("ENSG", 1:20) # 仮のEnsembl ID
colData <- DataFrame(condition = c("A", "A", "B", "B", "C"))
se <- SummarizedExperiment(assays = list(counts = counts), colData = colData)
# Ensemblデータベースからアノテーション情報を取得
ensembl <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")
annotation_data <- getBM(
attributes = c("ensembl_gene_id", "hgnc_symbol"),
filters = "ensembl_gene_id",
values = rownames(counts),
mart = ensembl
)
# 遺伝子IDをキーにアノテーションを追加(パイプラインを使わない形式)
row_data_df <- as.data.frame(rowData(se))
merged_data <- merge(row_data_df, annotation_data, by.x = "rowname", by.y = "ensembl_gene_id", all.x = TRUE)
rowData(se) <- DataFrame(merged_data)
# 結果を表示
print(rowData(se))
# 参考: パイプラインを使用したコード
# rowData(se) <- rowData(se) %>%
# as.data.frame() %>%
# left_join(annotation_data, by = c("rowname" = "ensembl_gene_id")) %>%
# DataFrame()
確認事項:
- Ensembl IDに基づいて、遺伝子シンボル情報が正しく統合されているか確認する。
演習3: シングルセルデータのバッチ結合(SingleCellExperiment)
- 2つの異なるバッチのシングルセルデータを作成し、それらを結合する。
cbind()を使って、SingleCellExperimentオブジェクトの結合が正しく行われるか確認する。
library(SingleCellExperiment)
# シングルセルRNA-seqデータの作成
expr_data1 <- matrix(rpois(20, lambda = 5), nrow = 5)
expr_data2 <- matrix(rpois(20, lambda = 5), nrow = 5)
# バッチ情報を含むサンプルアノテーションを作成
batch_info1 <- DataFrame(SampleID = paste0("Cell", 1:4), Batch = "Batch1")
batch_info2 <- DataFrame(SampleID = paste0("Cell", 5:8), Batch = "Batch2")
# SingleCellExperimentオブジェクトの作成
sce1 <- SingleCellExperiment(assays = list(counts = expr_data1), colData = batch_info1)
sce2 <- SingleCellExperiment(assays = list(counts = expr_data2), colData = batch_info2)
# バッチ間のデータ結合
combined_sce <- cbind(sce1, sce2)
# サンプル情報と発現データを確認
print(colData(combined_sce))
print(assay(combined_sce, "counts"))確認事項:
- バッチ間でシングルセルデータが正しく結合されているか確認する。また、
colDataにバッチ情報が保持されているかも確認する。
データの変換・要約
概要
ここまでに、データ構造の理解、ベクトル操作、結合操作について学んだ。次にデータの変換・要約について学ぶ。データの変換・要約処理は、列方向のサンプルごとの特徴あるいは行方向の特徴量(遺伝子)ごとの特徴を把握する目的で、総和を計算したり、スケールに依存しない偏りを評価するためのZスコアに変換したり、簡単な特徴量として分散や平均を計算したりする。
バイオインフォマティクスにおける変換・要約の応用
バイオインフォマティクス解析において、このような計算を行う場面は主に2つある。一つはフィルタリングのためのスコアを計算することであり、もう一つは、可視化のための統計データを作成するためである。
フィルタリングの役割と重要性
フィルタリングは、重要ではない特徴を解析から除外する目的で行われるものである。遺伝子発現データなどの膨大なデータでは、重要な特徴を持つ遺伝子やサンプルを選別するために行われる。たとえば、遺伝子発現の総和や平均、あるいは分散・標準偏差を計算し、一定の基準を満たさない遺伝子やサンプルを除外することが一般的である。フィルタリングによってデータが軽量化されたり、偽陽性の可能性を可能な限り排除して、解析やモデル構築を効率化する効果がある。
可視化と統計データの利用
また、可視化は、データ解析のさまざまなステップで重要な役割を果たす。多くのパッケージには、標準的な可視化関数が用意されているが、これらは探索的にデータを調査するには十分でない場合が多い。そのため、自分で必要な情報をデータ構造から抽出し、適切な可視化手法を選択して実装できるように習熟することが望ましい。
品質管理とバッチ効果の可視化
品質管理において異常なサンプルを特定するためには、いくつかのアプローチがある。例えば、低次元空間にデータを射影して散布図を作成し、外れ値を視覚的に確認する方法や、Zスコアに基づくヒートマップを用いて遺伝子発現レベルを可視化する方法などが挙げられる。これにより、サンプル間の発現パターンの違いや、異常な発現を示すサンプルを複数の観点から検証できる。
また、バッチ効果の検証においても、低次元空間における散布図を使用して、異なるバッチに由来するサンプルのグループ構造を視覚化する方法が有効である。さらに、各サンプルにおける全遺伝子の発現量をボックスプロットや密度分布で表し、全体の発現傾向を確認することもよく行われる。
これらの可視化手法の多くは、ggplot2やpheatmapといった統計的変換を含むパッケージによって包括的に扱うことができる。しかし、細かい可視化の調整や複合的な図を作成する際には、自らデータを変換し、要約処理を行ってカスタマイズすることが少なくない。
効率的なデータ操作と要約の重要性
これらの処理の多くは、解析のプロセスのなかで、表形式のデータ構造に対する操作であることが一般的で、データフレームもしくは行列に対して行われれることが多い。そのため、これらのデータ構造に対する処理・要約を効率的に行える操作を理解することは重要である。
使用する主要な関数とパッケージ
データの変換・要約に関しては、apply() 関数や
dplyr
パッケージを使用することで、データ構造全体にわたる計算を効率的に行うことができる。これにより、列ごとの要約や行ごとの変換をシンプルに書くことができ、for
ループに頼らずとも簡潔なコードで目的を達成できる。以下に、よく使用される手法と関数について説明する。
1. apply() 関数
apply()
関数は、行列やデータフレームに対して行や列ごとに関数を適用するために使われる。行ごとの合計、列ごとの平均などを計算する際に非常に便利である。
例1: 行ごとの合計を計算
# 行列データを作成
data <- matrix(rnorm(20), nrow = 5)
# 行ごとの合計を計算
row_sums <- apply(data, 1, sum)
print(row_sums)この例では、apply(data, 1, sum)
によって、行ごとの合計が計算されている。1
は行方向を示しており、sum 関数が各行に適用される。
例2: 列ごとの平均を計算
# 列ごとの平均を計算
col_means <- apply(data, 2, mean)
print(col_means)ここでは、2
を指定することで列ごとの平均が計算される。apply()
関数は汎用的なツールとして、さまざまな関数をデータ構造に適用するために使われる。他にもリスト要素ごとに適用可能なlapply()やsapply()もある。これらはapplyファミリーと呼ばれており、apply/sapply/lapply以外は使用頻度は多くはないが、その他について関数とその使い方、有用な場面を表にまとめておく。
| 関数名 | 説明 | 使い方の例 | 主な用途と有用な場面 |
|---|---|---|---|
| apply() | 行列やデータフレームに対して、行や列ごとに関数を適用する。 | apply(data, 1, sum) で行ごとの合計を計算 |
行列の各行または各列に同じ処理を施す場合。例: 行列の行ごとの合計、列ごとの平均を計算する。 |
| lapply() | リストやベクトルに対して関数を適用し、リストとして結果を返す。 | lapply(my_list, mean) でリスト内の平均を計算 |
リストの各要素に対して同じ処理を行い、その結果をリスト形式で保持したい場合。例: リスト内の各ベクトルに対して集計処理を行う。 |
| sapply() | lapply()
に似ているが、結果を可能であればベクトルや行列として返す。 |
sapply(my_list, mean) でベクトルとして結果を返す |
lapply()
と同様の操作を行いつつ、より簡潔な形式(ベクトルや行列)で結果を取得したい場合。例:
データフレームの各列に対して平均を計算し、ベクトルとして返す。 |
| vapply() | sapply()
に似ているが、返される値の型を指定できる。 |
vapply(my_list, mean, numeric(1)) で型を指定 |
sapply()
のように結果をベクトルや行列として返しつつ、型を保証したい場合。例:
処理の結果が特定の型(数値、文字列)であることを確実にしたい場合。 |
| mapply() | 複数のベクトルやリストに対して、要素ごとに関数を適用する。 | mapply(sum, 1:5, 6:10) で要素ごとの合計を計算 |
複数の引数を持つ関数に対して、複数のベクトルやリストを並行して処理したい場合。例: 2つのベクトルの要素ごとに処理を行う。 |
| tapply() | ベクトルの値をグループごとに分割し、各グループに関数を適用する。 | tapply(scores, group, mean)
でグループごとの平均を計算 |
データをグループ化して、そのグループごとに関数を適用したい場合。例: サンプルをグループごとに分け、その中で平均や合計を計算する。 |
| rapply() | リストのネストされた要素に対して再帰的に関数を適用する。 | rapply(nested_list, sqrt)
でネスト内の要素に平方根を適用 |
ネストされたリストに対して再帰的に処理を行いたい場合。例: 複雑なリスト構造内のすべての数値要素に対して操作を行う。 |
| eapply() | 環境に含まれる全てのオブジェクトに関数を適用する。 | eapply(env, mean)
で環境内のオブジェクトに処理を適用 |
環境内のすべてのオブジェクトに対して関数を適用したい場合。例: 環境内のデータフレームに対して一括処理を行う。 |
また、applyファミリーで実装可能ではあるが、行・列の総和や最大値、z-scoreなどより簡便にパッケージ関数として提供されているものがある。それらについてもまとめておく。これらの関数は、行列やデータフレームに対して行または列ごとの集計や変換を効率的に行うために利用される。特に、バイオインフォマティクス解析のような大規模データ処理においては、各遺伝子やサンプルごとの特徴量を計算したり、フィルタリングを行う際に重要である。
行方向・列方向の操作に使える関数一覧
| 関数 | 説明 | 使用例 |
|---|---|---|
rowSums() |
各行の合計値を計算する | rowSums(matrix_data) |
colSums() |
各列の合計値を計算する | colSums(matrix_data) |
rowMeans() |
各行の平均値を計算する | rowMeans(matrix_data) |
colMeans() |
各列の平均値を計算する | colMeans(matrix_data) |
scale() |
行列やデータフレームの列を標準化(Zスコア)する | scale(matrix_data) |
t() |
行列の転置を行う(行と列を入れ替える) | t(matrix_data) |
rowMin() |
各行の最小値を計算する(matrixStats
パッケージが必要) |
rowMin(matrix_data) |
colMin() |
各列の最小値を計算する(matrixStats
パッケージが必要) |
colMin(matrix_data) |
rowMax() |
各行の最大値を計算する(matrixStats
パッケージが必要) |
rowMax(matrix_data) |
colMax() |
各列の最大値を計算する(matrixStats
パッケージが必要) |
colMax(matrix_data) |
rowVars() |
各行の分散を計算する(matrixStats
パッケージが必要) |
rowVars(matrix_data) |
colVars() |
各列の分散を計算する(matrixStats
パッケージが必要) |
colVars(matrix_data) |
rowMedians() |
各行の中央値を計算する(matrixStats
パッケージが必要) |
rowMedians(matrix_data) |
colMedians() |
各列の中央値を計算する(matrixStats
パッケージが必要) |
colMedians(matrix_data) |
使用場面の例
rowSums()/colSums():- 遺伝子発現データに対して、各遺伝子ごと(行)の発現総量や各サンプルごと(列)の発現総量を計算する際に便利。
scale():- データを標準化して解析に適したスケールに変換する際に使用(Zスコア変換など)。
matrixStats関連の関数:matrixStatsパッケージには、行列に対する分散や中央値、最小値、最大値などの計算を効率的に行う関数が豊富に用意されているため、特に大規模な行列データに対して使用することが多い。
使用例
行列データの標準化
行列データを標準化する場合、各列ごとに平均を引いて標準偏差で割るZスコア変換を行うことがよくある。この操作も
apply() 関数や scale()
関数を使って簡単に実装できる。
例3: 列ごとにZスコアを計算
# 行列データを標準化
scaled_data <- scale(data)
print(scaled_data)scale()
関数は、デフォルトで各列の平均を0、標準偏差を1に標準化してくれる。これにより、異なるスケールのデータを統一されたスケールに変換することができる。
行ごと・列ごとの特徴量の抽出とフィルタリング
変換や要約の目的で、フィルタリングを行うことも重要である。例えば、分散が低い遺伝子をフィルタリングして除外したり、特定の条件を満たすサンプルだけを抽出したりする。
例4: 分散が低い遺伝子をフィルタリング
# 各行の分散を計算
variances <- apply(data, 1, var)
# 分散がしきい値以上の行を選択
filtered_data <- data[variances > 0.5, ]
print(filtered_data)この例では、apply()
関数を使って各行の分散を計算し、しきい値を設定して分散が一定以上の遺伝子だけを抽出している。
複数の変換ステップの適用
解析の際には、複数の変換ステップを組み合わせて適用することがよくある。例えば、まずフィルタリングを行い、その後標準化を行うといった流れが一般的である。
例5: フィルタリングと標準化の組み合わせ
# 分散フィルタリング後、標準化
filtered_scaled_data <- scale(filtered_data)
print(filtered_scaled_data)2. dplyr パッケージによる要約操作
dplyr パッケージでは、summarise() や
mutate()
を使って、複雑な変換や要約処理を行うことが可能である。また、group_by()を組み合わせることで、条件ごとの集約や変換も効率的に処理できる。この機能により、データをより可読性の高い形式で要約するだけでなく、複雑な条件付き処理にも対応できる。
例6: 列ごとの特徴量を計算
library(dplyr)
# サンプルデータフレームを作成
df <- data.frame(
ID = 1:5,
Score = c(85, 92, 78, 90, 87),
Group = c("A", "B", "A", "B", "A")
)
# グループごとの平均スコアを計算
mean_score <- df %>%
group_by(Group) %>%
summarise(mean = mean(Score))
print(mean_score)この例では、dplyrのgroup_by()とsummarise()を使って、Score列をGroupでグループ化し、それぞれのグループの平均を計算している。複数の条件付き処理に対応し、効率的に計算できる点がapplyファミリーと比べて優れている。
例7: 新しい列を追加してデータを変換
# Zスコアを計算して新しい列を追加
df <- df %>%
mutate(Z_Score = (Score - mean(Score)) / sd(Score))
print(df)この例では、mutate()関数を用いてScoreのZスコアを計算し、新たな列としてデータフレームに追加している。このように、変換や新しい特徴量の追加を行う際にも、dplyrの方が簡潔かつ可読性の高いコードが書ける。
例8: 条件付きのデータ変換と要約
# Group "A" のみにZスコアを計算して追加
df <- df %>%
group_by(Group) %>%
mutate(Z_Score = if_else(Group == "A", (Score - mean(Score)) / sd(Score), NA_real_))
print(df)この例では、mutate()とif_else()を組み合わせることで、特定の条件(ここではGroupが”A”であるかどうか)に基づいて、Zスコアの計算を行っている。dplyrを使用すると、このような条件付きの変換処理もシンプルに記述できるため、複雑な条件付き処理においてapplyファミリーよりも明らかに優位性がある。
3. long/wide型のデータ形式変換
ggplot2のような可視化パッケージでデータをプロットする際や、複数のカテゴリデータを比較する場合に、しばしば表データの形式変換(long型⇔wide型)が必要な場面に遭遇する。ggplot2ではデータフレームを入力とする関数からなっているが、データフレームを適宜変換する操作には慣れが必要である。この際に、dplyr
と tidyr
パッケージを組み合わせることで、データの形式変換(long型⇔wide型)が簡単に行える。
long型とwide型のデータ形式変換の例
ここでは、遺伝子発現量に加え、disease/healthy(疾患状態)、age(年齢)、sex(性別)といったカテゴリ変数を含むデータセットを例に、dplyrとtidyrを使用してlong型とwide型のデータ形式の変換を行う。
まず、例として次のようなwide型の遺伝子発現データを考える。ここでは、異なる条件(健康状態、年齢、性別)における複数の遺伝子発現量が記録されている。
wide型データ例:
| SampleID | GeneA | GeneB | GeneC | Disease_Status | Age | Sex |
|---|---|---|---|---|---|---|
| 1 | 5.4 | 3.2 | 7.1 | Healthy | 45 | Male |
| 2 | 6.1 | 2.8 | 6.9 | Disease | 60 | Female |
| 3 | 5.9 | 3.5 | 7.4 | Disease | 50 | Male |
| 4 | 5.2 | 3.1 | 7.0 | Healthy | 38 | Female |
このデータセットには3つの遺伝子(GeneA, GeneB, GeneC)の発現量が記録されており、各サンプルについて健康状態、年齢、性別の情報が含まれている。
wide型からlong型への変換
long型に変換することで、各サンプルの遺伝子発現データがより柔軟に扱えるようになる。pivot_longer()を使用して、遺伝子発現の情報を一列にまとめ、Gene列に遺伝子名を、Expression列に発現量を格納する形式にする。
library(dplyr)
library(tidyr)
# サンプルデータ
df <- data.frame(
SampleID = c(1, 2, 3, 4),
GeneA = c(5.4, 6.1, 5.9, 5.2),
GeneB = c(3.2, 2.8, 3.5, 3.1),
GeneC = c(7.1, 6.9, 7.4, 7.0),
Disease_Status = c("Healthy", "Disease", "Disease", "Healthy"),
Age = c(45, 60, 50, 38),
Sex = c("Male", "Female", "Male", "Female")
)
# wide型からlong型への変換
long_df <- df %>%
pivot_longer(cols = starts_with("Gene"),
names_to = "Gene",
values_to = "Expression")
print(long_df)結果(long型データ):
| SampleID | Disease_Status | Age | Sex | Gene | Expression |
|---|---|---|---|---|---|
| 1 | Healthy | 45 | Male | GeneA | 5.4 |
| 1 | Healthy | 45 | Male | GeneB | 3.2 |
| 1 | Healthy | 45 | Male | GeneC | 7.1 |
| 2 | Disease | 60 | Female | GeneA | 6.1 |
| 2 | Disease | 60 | Female | GeneB | 2.8 |
| 2 | Disease | 60 | Female | GeneC | 6.9 |
| 3 | Disease | 50 | Male | GeneA | 5.9 |
| 3 | Disease | 50 | Male | GeneB | 3.5 |
| 3 | Disease | 50 | Male | GeneC | 7.4 |
| 4 | Healthy | 38 | Female | GeneA | 5.2 |
| 4 | Healthy | 38 | Female | GeneB | 3.1 |
| 4 | Healthy | 38 | Female | GeneC | 7.0 |
この形式に変換することで、複数の遺伝子発現量を一列にまとめ、遺伝子名と発現量を別々に扱えるようになる。この形式は、ggplot2でカテゴリ別に遺伝子発現量をプロットする際に非常に便利である。
long型からwide型への変換
次に、再びwide型に戻す例を示す。pivot_wider()を使って、Gene列を新しい列として展開し、それぞれの遺伝子ごとに発現量を表示する。
# long型からwide型へ変換
wide_df <- long_df %>%
pivot_wider(names_from = Gene, values_from = Expression)
print(wide_df)結果(wide型データ):
| SampleID | Disease_Status | Age | Sex | GeneA | GeneB | GeneC |
|---|---|---|---|---|---|---|
| 1 | Healthy | 45 | Male | 5.4 | 3.2 | 7.1 |
| 2 | Disease | 60 | Female | 6.1 | 2.8 | 6.9 |
| 3 | Disease | 50 | Male | 5.9 | 3.5 | 7.4 |
| 4 | Healthy | 38 | Female | 5.2 | 3.1 | 7.0 |
この操作により、再び各遺伝子が個別の列として表示され、分析や確認がしやすいwide形式に戻すことができる。
long/wide形式変換の意義
- long型:
各サンプルの複数の遺伝子発現を1列にまとめることで、
ggplot2などで遺伝子ごとの発現パターンを可視化する際に便利。 - wide型: データの確認や簡単な要約統計を計算する際には、wide型が適している。
これらの形式変換を活用することで、遺伝子発現データを柔軟に操作し、解析や可視化の目的に応じて適切な形式でデータを扱うことができる。
applyファミリーとdplyrの比較
データの変換・要約処理は、データ解析の中で重要なステップであり、データの特徴を効率的に抽出するために用いられる。apply()
関数や dplyr
パッケージを駆使することで、簡潔かつ効率的にデータ変換が行える。適切な関数を選択することで、複雑な操作をシンプルに記述し、解析をスムーズに進めることができるようになる。
apply ファミリーの関数と dplyr
は、どちらもデータ操作に非常に有用だが、使い分けの基準は操作するデータ構造や操作の性質に応じて異なる。それぞれの特徴を考慮して使い分ける基準を以下に示す。
データ構造に基づく使い分け
applyファミリー- 行列やリスト、ベクトル の操作に強みがある。特に行列やリストに対して関数を行ごと・列ごとに適用する際に有効。
- リストや行列に対して再帰的に操作を行う場合や、特定の型を保証しながら関数を適用する場合に役立つ。
dplyr- データフレーム(特に大規模データフレーム) を効率的に操作するために設計されている。データフレームの列に対して関数を適用したり、データの要約、変換、フィルタリングを行うのに便利。
group_by()やsummarise()を使ったグループ化操作や、特定の条件に基づくデータの操作(列の追加、更新など)が得意である。
操作の性質に基づく使い分け
applyファミリー- 行や列に沿った単純な計算(例えば、行ごとの和、列ごとの平均など)を行う場合は
apply()が有効。 - リストやネストされたデータ構造に対して関数を適用する場合や、ベクトルを使った並列処理には
lapply()やsapply()が便利である。 - リストや複数のベクトルの要素ごとの操作には
mapply()を使用。 - ベクトルや行列の値をグループごとに集計する場合には
tapply()が適している。
- 行や列に沿った単純な計算(例えば、行ごとの和、列ごとの平均など)を行う場合は
dplyr- 複数のステップからなるデータ変換や、パイプライン処理(
%>%)でデータの操作を直感的に記述したい場合に強力である。 - 可読性の高いコード を必要とする場面や、特定の列に対してフィルタリングや集約を行いたい場合に有用である。
- データフレーム内でのグループ化操作(
group_by())や要約統計量の計算(summarise())、条件付き列の追加や更新(mutate())にはdplyrが最適である。 - パフォーマンス
を考慮したい場合、大規模データセットに対して
dplyrを使うと、バックエンドにdata.tableを使用したりして効率的な計算が可能だ。
- 複数のステップからなるデータ変換や、パイプライン処理(
複雑な処理と再現性に基づく使い分け
applyファミリー- 単純な操作を手早く行いたい場合や、コードの中で他の関数の引数として関数適用を使いたい場合に向いている。
- 簡単な集計や変換(行列やリストの処理)には
apply()ファミリーがシンプルかつ効果的。
dplyr- 複数の変換ステップや操作を組み合わせる際には、
dplyrの方が処理の流れを視覚的に捉えやすい。 - 再現性やコードの可読性を重視する場合、
dplyrのパイプライン (%>%) を使った方がコードが明瞭になる。
- 複数の変換ステップや操作を組み合わせる際には、
まとめると、
applyファミリー は、行列やリストに対する繰り返し操作、再帰的な処理やリスト操作に向いている。特にリストやベクトルに対して個別に関数を適用する際にシンプルで便利。dplyrは、データフレームの操作、フィルタリング、要約統計量の計算に向いており、コードの可読性や処理の流れを直感的に表現できる。特に、グループ化操作やパイプライン処理を使いたい場合に強力。
複雑なオブジェクトに対する処理について
また、SummarizedExperiment や
SingleCellExperiment
オブジェクトに対しても、データ構造からそれぞれのコンポーネントに対して、さまざまなデータ変換・要約処理が可能である。ただし、SummarizedExperiment
や SingleCellExperiment
そのものは、発現データやアノテーション情報の管理を行うためのデータ構造を提供しているに過ぎない。
SummarizedExperiment や
SingleCellExperimentのアッセイ情報に対する変換・要約処理は、基本的にはユーザーが自ら行うものであり、これらのデータ構造に対して行う変換や要約処理(例えば、Zスコア変換、平均や分散の計算、フィルタリングなど)は、Rの基本関数や他のパッケージの関数を利用して行うことが一般的である。
ただし、SummarizedExperiment や
SingleCellExperimentを用いた変動解析のように、特定のワークフローに必要な処理については、Bioconductorの関連パッケージ内で行われる。例えば、RNA-seqデータの解析では
DESeq2 や edgeR
のようなパッケージが提供する関数を用いることで、発現データの変換・要約処理が自動的に行われる。あくまでもカスタムのフィルタリング処理条件や可視化において必要な操作として理解しておくとよい。
発現データの行列に対する変換処理
発現データ(assays
スロットに格納されている行列)に対して、通常の行列操作が可能である。例えば、遺伝子ごとの分散や平均の計算、Zスコアへの変換など、基本的な統計的処理を行うことができる。
例: 行ごとの平均と分散を計算
# SummarizedExperimentオブジェクトの発現データを取得
counts <- assay(se)
# 遺伝子ごとの平均と分散を計算
gene_means <- rowMeans(counts)
gene_vars <- apply(counts, 1, var)
print(gene_means)
print(gene_vars)この例では、SummarizedExperiment の assay()
関数を使って遺伝子発現データを取り出し、各遺伝子の平均と分散を計算している。apply()
関数や rowMeans()
など、通常の行列操作がそのまま適用可能である。
可視化
可視化とは、データ解析において、あらゆる文脈でデータと対話するために不可欠な技術である。可視化は単に形式的なものや、見栄えの良い図を作成するためだけのものではなく、データを見る人が正確で深い理解を得るためのプロセスでもある。可視化は、常にデータの本質を視覚的に表現し、見る人が誤解をしないようにデザインされるべきであり、意図的に偏った解釈に誘導するようなものではあってはならない。とはいえ、データのすべての情報を無制限に表示しても、逆にその意図が伝わらず、重要な情報が埋もれてしまうことがある。
「良い」可視化に絶対的な基準が存在するわけではないが、少なくとも満たすべき基本的な要素がある。それは、視覚的に伝えたいメッセージを明確に表現し、適切な方法で情報を強調することである。特に、可視化の目的に応じて、選択するグラフの種類や、使用するツール(たとえば、RやPythonのパッケージ)が変わる。可視化の効果を最大限に引き出すためには、適切な可視化手法を選び、それが伝えたい仮説や結論に沿ったものであるかを常に意識する必要がある。
そこで、まずは可視化の主な目的について理解を深め、その後で利用可能なさまざまな可視化手法を概観する。次に、目的に応じてどのように可視化のタイプを選び、使用するツールやパッケージを使い分けるかについて学んでいく。最終的には、可視化のプロセスを通じて、解析結果をより効果的に伝えられるようにするための考え方やスキルを身につけることが目標である。
1. 可視化の目的と役割
可視化の目的の多様性
可視化の目的は一様ではなく、データ解析の文脈や目的に応じて、その役割が異なる。大きく分けて次の3つの目的がある。
- データの傾向を直感的に理解するため(探索的解析):
- データの可視化は、解析の初期段階で行われることが多い。大量のデータの中から重要なパターンや異常値、トレンドなどを発見するために、まずはデータを視覚化して全体像を把握する。これにより、どのような仮説を立てるべきか、またはどの部分に着目して解析を進めるべきかが明らかになる。探索的データ解析(EDA)では、シンプルで迅速な可視化手法が重視され、
ggplot2やbase plotのような柔軟なツールが活躍する。
- データの可視化は、解析の初期段階で行われることが多い。大量のデータの中から重要なパターンや異常値、トレンドなどを発見するために、まずはデータを視覚化して全体像を把握する。これにより、どのような仮説を立てるべきか、またはどの部分に着目して解析を進めるべきかが明らかになる。探索的データ解析(EDA)では、シンプルで迅速な可視化手法が重視され、
- 仮説を検証し、重要な情報を強調するため(説明的解析):
- 仮説に基づく解析を行う際、可視化はデータの裏付けを視覚的に示すための手段となる。たとえば、グループ間の比較や相関関係を明示するために、棒グラフ、散布図、箱ひげ図などを用いて、特定のデータポイントやトレンドを強調する。この段階では、解釈を容易にするためにデータのノイズを除去したり、特定の変数に焦点を絞った可視化が求められる。
- 論文やプレゼンテーション用に高品質な可視化を作成するため(発表・公表用):
- 最終的にデータを他者に伝える際、可視化はデータの要点を明確に伝えるための強力なツールとなる。論文やプレゼンテーションで使用する図は、視覚的な美しさや分かりやすさが重要であり、配色、ラベル付け、凡例の配置、解像度などにも注意が必要である。ここでは、発表目的に合わせてデザインされた高品質な図が求められ、
ggplot2やplotlyなどのツールを活用して、データの意図が正確に伝わるようにする。
- 最終的にデータを他者に伝える際、可視化はデータの要点を明確に伝えるための強力なツールとなる。論文やプレゼンテーションで使用する図は、視覚的な美しさや分かりやすさが重要であり、配色、ラベル付け、凡例の配置、解像度などにも注意が必要である。ここでは、発表目的に合わせてデザインされた高品質な図が求められ、
下流解析における具体的役割
生物学的なハイスループット測定における可視化の本質は、膨大で多次元なデータを視覚的に解釈可能にすることで、実験結果や仮説を統合的に理解するための不可欠な手段である。以下に、可視化が果たす役割とその本質的な目的を、生物学的な視点から整理して説明する。
1. 技術的バイアスやバッチ効果によるグループ構造の検出
ハイスループット測定データは、技術的なバイアスやバッチ間の違い(バッチ効果)により、データ内に意図しないパターンが生じることがある。可視化を用いることで、技術的要因による影響を検出し、これを補正するための手がかりを得ることができる。
- 例: PCAやUMAPプロットで、サンプルが測定バッチごとに分離しているかを確認し、技術的バイアスの存在を視覚的に捉える。
2. 生物学的なグループ構造の発見
可視化のもう一つの重要な目的は、データ内に潜む生物学的なグループ構造を明らかにすることである。ハイスループット測定データは、異なる条件や生物学的状態(例えば健康な個体と疾患の個体)を含んでおり、これらの違いを可視化することで、生物学的な特徴を浮かび上がらせる。
- 例: UMAPプロットで異なる疾患状態に応じてサンプルがグルーピングされることで、特定の分子パターンに基づいた疾患サブタイプの発見が可能になる。
3. 品質管理
ハイスループットデータの可視化は、データの品質をチェックし、解析の信頼性を確保するための重要なステップである。異常値やノイズを検出し、データが適切に収集され、解析可能な状態であるかを確認することができる。
- 例: ボックスプロットやヒートマップを使って、特定のサンプルや遺伝子の値が極端に異常でないかを確認し、不適切なデータを排除する。
4. 各処理・解析ステップごとの方法の評価
データ解析の各ステップ(フィルタリング、正規化、モデル適用など)における結果を可視化することで、選択した解析手法が適切であったかを評価する。例えば、正規化前後のデータを比較して、処理の効果を視覚的に確認できる。
- 例: RNA-seqデータの正規化前後の分布をヒストグラムで比較することで、サンプル間のバイアスが補正されたかを確認する。
5. 仮説の確からしさ(実験・介入効果の可視化)
可視化は、実験や介入の効果を確認し、仮説がデータによって支持されているかを検証する手段としても用いられる。具体的には、条件間での発現変動や、介入後の変化を視覚化することで、仮説の妥当性を確かめる。
- 例: 発現変動をボルケーノプロットで可視化し、実験条件により有意に発現が変動している遺伝子群を特定する。
6. システム生物学的な分子間の複雑な関係性(相互作用)、分子モジュールの発見
システム生物学の文脈では、分子間の相互作用やネットワーク構造を可視化することが、複雑な分子メカニズムを理解する鍵となる。これにより、分子モジュールや重要なネットワークハブを特定し、分子機構の全体像を把握することができる。
- 例: ネットワーク解析に基づく分子間相互作用を可視化し、相互作用の強度やネットワークの中心にいる重要な分子を特定する。
7. データ全体の俯瞰
膨大なハイスループットデータを一望できる可視化は、データの全体像を把握し、どの部分に着目すべきかを見極めるために重要である。これにより、データ解析の方向性を定めたり、仮説を生み出すためのインスピレーションを得ることができる。
- 例: ヒートマップを使って、発現パターンが似ている遺伝子群やサンプル群を全体的に把握する。
以下に、上記の7つの事例について、Rで実際に可視化を行うための演習コード例を示します。各事例で使用するグラフや可視化方法に沿って、必要なRパッケージを使ってデータを視覚化します。これらの演習を通じて、可視化の目的やその適用方法を実際に体験していきましょう。
演習1: 技術的バイアスやバッチ効果の検出
目的: PCAプロットを用いて、技術的バイアス(バッチ効果)がサンプル間にあるかどうかを確認する。
# パッケージをロード
library(ggplot2)
library(DESeq2)
# サンプルのデータ作成 (例: RNA-seqデータ)
set.seed(123)
counts <- matrix(rpois(1000, lambda = 10), ncol = 10)
batch <- factor(rep(c("Batch1", "Batch2"), each = 5))
colData <- data.frame(batch = batch)
# DESeq2オブジェクトを作成
dds <- DESeqDataSetFromMatrix(countData = counts, colData = colData, design = ~batch)
# 正規化
vsd <- vst(dds)
# PCAプロット
pcaData <- plotPCA(vsd, intgroup = "batch", returnData = TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
ggplot(pcaData, aes(PC1, PC2, color = batch)) +
geom_point(size = 3) +
xlab(paste0("PC1: ", percentVar[1], "% variance")) +
ylab(paste0("PC2: ", percentVar[2], "% variance")) +
ggtitle("PCA plot with batch effect")確認事項: サンプルがバッチごとに分かれてクラスタリングされているかどうかを確認し、技術的バイアスが存在するかを検討する。
演習2: 生物学的なグループ構造の発見
目的: UMAPプロットを用いて、生物学的なグループ(例: 健康 vs. 疾患)を発見する。
# パッケージをロード
library(ggplot2)
library(umap)
# サンプルデータ作成
set.seed(123)
data <- matrix(rnorm(2000), nrow = 200, ncol = 10)
group <- factor(rep(c("Control", "Disease"), each = 5))
# UMAPを実行
umap_result <- umap(data)
# 結果をプロット
umap_df <- data.frame(UMAP1 = umap_result$layout[,1],
UMAP2 = umap_result$layout[,2],
group = group)
ggplot(umap_df, aes(UMAP1, UMAP2, color = group)) +
geom_point(size = 3) +
ggtitle("UMAP plot showing biological groups")確認事項: UMAPプロットでサンプルが健康(Control)と疾患(Disease)のグループで明確に分かれているかを確認する。
演習3: 品質管理のための可視化
目的: ボックスプロット、PCAに基づく主成分スコアの散布図、ヒートマップを用いて、サンプル間で異常なデータポイントがないかを確認する。
# パッケージをロード
library(ggplot2)
library(DESeq2)
# サンプルデータ作成(通常サンプルと異常サンプルを含む)
set.seed(123)
counts <- matrix(rpois(900, lambda = 100), ncol = 9) # 通常サンプル
abnormal_sample <- rpois(100, lambda = 500) # 異常サンプル(外れ値)
counts <- cbind(counts, abnormal_sample) # 異常サンプルを追加
colnames(counts) <- paste0("Sample", 1:10)
colData <- data.frame(condition = factor(rep(c("A", "B"), each = 5)))
# DESeq2オブジェクトを作成
dds <- DESeqDataSetFromMatrix(countData = counts, colData = colData, design = ~ condition)
# ボックスプロットで各サンプルのリード数の分布、バイアスを確認
boxplot(counts, main = "Boxplot with Abnormal Sample for Quality Control",
xlab = "Samples", ylab = "Counts", outline = TRUE)
# varianceStabilizingTransformation() を使用して正規化
vsd <- varianceStabilizingTransformation(dds)
# PCAプロットを作成
pcaData <- plotPCA(vsd, intgroup = "condition", returnData = TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
ggplot(pcaData, aes(PC1, PC2, color = condition)) +
geom_point(size = 3) +
xlab(paste0("PC1: ", percentVar[1], "% variance")) +
ylab(paste0("PC2: ", percentVar[2], "% variance")) +
ggtitle("PCA Plot with Abnormal Sample for Quality Control")
確認事項: 異常に高いまたは低いサンプルがないか、サンプル間でバイアスがないかを確認する。
演習4: 各解析ステップの評価
目的: 正規化前後のRNA-seqデータのヒストグラムを比較し、正規化の効果を確認する。
# パッケージをロード
library(ggplot2)
library(DESeq2)
# サンプルデータ作成 (RNA-seqデータ)
set.seed(123)
counts <- matrix(rpois(1000, lambda = 10), ncol = 10)
# DESeq2オブジェクトを作成
dds <- DESeqDataSetFromMatrix(countData = counts, colData = data.frame(condition = factor(rep(c("A", "B"), each = 5))), design = ~ condition)
# 正規化前のヒストグラム
par(mfrow = c(1, 2))
hist(log2(counts + 1), breaks = 50, main = "Before normalization", xlab = "Log2(counts + 1)")
# varianceStabilizingTransformation()を使用して正規化
vsd <- varianceStabilizingTransformation(dds)
# 正規化後のヒストグラム
hist(assay(vsd), breaks = 50, main = "After normalization", xlab = "VST counts")
確認事項: 正規化前後の分布を確認し、正規化によりデータが整ったかどうかを判断する。
演習5: 仮説の確からしさ(発現変動の可視化)
目的: ボルケーノプロットで発現変動遺伝子を視覚化し、実験の結果を確認する。
# パッケージをロード
library(ggplot2)
library(DESeq2)
# サンプルデータ作成
set.seed(123)
dds <- makeExampleDESeqDataSet()
# 発現変動解析
dds <- DESeq(dds)
res <- results(dds)
# 欠損値の処理
res$log2FoldChange[is.na(res$log2FoldChange)] <- 0
res$pvalue[is.na(res$pvalue)] <- 1
# 遺伝子名をラベルとして追加
res$gene <- rownames(res)
# ボルケーノプロット
ggplot(res, aes(x = log2FoldChange, y = -log10(pvalue))) +
geom_point(alpha = 0.5) +
ggtitle("Volcano Plot") +
xlab("Log2 Fold Change") +
ylab("-Log10 P-value") +
geom_hline(yintercept = -log10(0.05), linetype = "dashed", color = "red") +
# 閾値を満たす遺伝子にラベルを追加
geom_text(
data = subset(res, abs(log2FoldChange) > 2 & pvalue < 0.05),
aes(label = gene),
vjust = 1.5,
color = "blue",
size = 3
)
確認事項: 有意に発現変動している遺伝子がボルケーノプロット上で強調されているか確認する。
演習6: 分子間の相互作用の可視化
目的: ネットワーク解析に基づく分子間の相互作用を可視化する。
# パッケージをロード
library(igraph)
# サンプルネットワークデータ作成
nodes <- data.frame(name = paste0("Gene", 1:10))
edges <- data.frame(from = sample(nodes$name, 10, replace = TRUE),
to = sample(nodes$name, 10, replace = TRUE))
# グラフオブジェクトの作成
graph <- graph_from_data_frame(edges, vertices = nodes, directed = FALSE)
# ネットワークプロット
plot(graph, vertex.label = V(graph)$name, main = "Gene Interaction Network")確認事項: 分子間の相互作用を視覚化し、重要なノードや相互作用を発見できるか確認する。
演習7: データ全体の俯瞰
目的: ヒートマップを使用して、発現パターンを俯瞰し、遺伝子やサンプルの全体像を把握する。
# パッケージをロード
library(pheatmap)
# サンプルデータ作成
set.seed(123)
data <- matrix(rnorm(100), nrow = 10)
# ヒートマップを作成
pheatmap(data, cluster_rows = TRUE, cluster_cols = TRUE,
main = "Heatmap of Gene Expression")2. 可視化の基本的な要素
データ可視化を効果的に行うためには、データの構造に基づいて適切なグラフを選択することが重要である。データの構造や伝えたい情報に応じて異なるグラフを使用することで、視覚的にわかりやすく、情報を正確に伝えることができる。ここでは、データの可視化において基本的な要素である、グラフの選択、ラベルや軸の設計、およびカラースキームの選択について説明する。
データの構造に基づく可視化の選択
1変数データ(単一変数データ): 1変数データの可視化では、データの分布や要約統計量を視覚的に示すことが目的となる。例えば、データの広がりや極端な値(外れ値)を把握するためのグラフがよく用いられる。
- ヒストグラム:
データの分布を直感的に理解するための基本的なグラフ。各ビンの高さがその範囲に含まれるデータの頻度を表す。
- 使用例: 学生のテストスコアの分布を可視化。
- 箱ひげ図(Box Plot):
データの中央値、四分位範囲、最小値・最大値、外れ値を視覚的に示す。異常なデータポイントがあるかを確認するのに有効。
- 使用例: 遺伝子発現データの分布をサンプルごとに比較。
ポイント: 1変数データは、データの分布を視覚化するためのヒストグラムや、外れ値を含む全体像を確認するためのボックスプロットが効果的である。
- ヒストグラム:
データの分布を直感的に理解するための基本的なグラフ。各ビンの高さがその範囲に含まれるデータの頻度を表す。
2変数データ(2つの変数の関係): 2つの変数の間の関係性を示すグラフが必要となる。変数間の相関関係や、群ごとの違いを把握するために用いられる。
- 散布図(Scatter Plot):
2つの変数間の相関関係やトレンドを視覚化する。変数間の線形関係を評価する際に有効。
- 使用例: 遺伝子発現量と治療効果の相関を可視化。
- バイオリンプロット:
データの分布を示すが、密度プロットも併せて描かれるため、ボックスプロットに比べて分布の詳細が分かる。
- 使用例: 条件間での遺伝子発現の違いを可視化。
- 箱ひげ図(Box Plot):
サンプル間の分布を比較するために使われる。外れ値や中央値の比較が容易にできる。
- 使用例: サンプル間でのRNA-seqデータの分布の比較。
ポイント: 2変数データの可視化では、散布図で変数間の関係を把握し、バイオリンプロットや箱ひげ図を使って、群間の分布の違いを視覚的に示すことができる。
- 散布図(Scatter Plot):
2つの変数間の相関関係やトレンドを視覚化する。変数間の線形関係を評価する際に有効。
多変数データ(高次元データ): 多次元データでは、複数の変数を同時に扱う必要があり、次元削減やグループ化を行ってデータの構造を可視化する。
- ペアプロット(Pair Plot):
すべての変数の組み合わせに対して散布図を描く。変数間の関係性を複数視点から同時に確認できる。
- 使用例: 複数の遺伝子発現データセットの相関関係を確認。
- ファセットによる可視化:
特定のカテゴリでデータを分割し、複数の小さなグラフとして可視化する方法。グループごとの違いを把握するのに有効。
- 使用例: 複数のサンプルにおける遺伝子発現の分布を比較。
- 次元削減後の可視化(PCA, UMAP, t-SNEなど):
高次元データを2次元または3次元に圧縮して、データの全体像やグループ構造を把握する。
- 使用例: シングルセルRNA-seqデータをUMAPやt-SNEで次元削減し、細胞タイプごとにクラスタリングを視覚化。
ポイント: 多変数データの可視化では、次元削減技術(PCA, UMAP, t-SNE)を用いて高次元データを低次元に圧縮し、データの構造を明確にする。また、ペアプロットやファセットを使用することで、複数の変数間の関係を同時に可視化する。
- ペアプロット(Pair Plot):
すべての変数の組み合わせに対して散布図を描く。変数間の関係性を複数視点から同時に確認できる。
ラベルや軸の設計
- ラベルの明確さ:
軸ラベルやグラフのタイトルは、グラフが何を示しているのかを明確に伝える重要な要素である。ラベルは簡潔でありながら、必要な情報を含むべきである。
- 例: 遺伝子発現量 vs. サンプル条件のグラフには、「遺伝子発現量 (Log2)」や「サンプルグループ」などのラベルを付ける。
- 軸のスケールと目盛り: データの範囲に応じた適切なスケールを選ぶ。例えば、データが対数的に広がっている場合は、軸を対数スケールにすることが望ましい。また、目盛りは読みやすく、間隔が均等であることが重要である。
カラースキームの選択
- 見やすさと情報伝達のバランス:
カラースキームはデータの情報を強調するために使われる。特に、カテゴリ変数や連続変数を表現する際には、色が意味するところを適切に表現する必要がある。
- 例: 条件Aと条件Bを示すために、コントラストのある色(例えば、青と赤)を選択する。
- カラーブラインド対応:
色覚に障害のある人々にも配慮し、カラースキームを選ぶ際にはカラーブラインド対応の配色(例:
viridis)を使用することが推奨される。
可視化の際には、どのグラフを使用するかの選択が非常に重要であり、それはデータの構造や、何を伝えたいかという意図に大きく依存する。適切なグラフを選択し、軸やカラースキームを工夫することで、データの全体像や重要なポイントを視覚的に強調できる。
3. コードによる実装例: 探索的解析と説明的解析
データ解析において、可視化は迅速な探索的解析と、詳細な説明的解析の両方で重要な役割を果たす。以下では、シンプルな可視化と、高度な可視化パッケージによる詳細なカスタマイズ(例えばggplot2)ををい分ける方法を紹介する。
- 迅速なデータ探索のためのシンプルな可視化:
plot(),hist(),boxplot()などのベースRによる手早い可視化- 簡単にデータの全体像を掴むために使用
- 高度な可視化パッケージによる詳細なカスタマイズ:
ggplot2を使ったカスタマイズ可能なグラフ作成- 軸ラベル、色分け、凡例の位置、ファセットを使った可視化の細かい調整
- 発表・論文用に耐えうる品質を作成
迅速なデータ探索のためのシンプルな可視化
ベースRには、シンプルで効率的に可視化できる関数が多く用意されている。これらを使うことで、解析の初期段階でデータの全体像を素早く把握することが可能である。
- ベースRによる可視化の例
plot(): 変数間の関係を確認する散布図など、基本的なグラフ作成が可能。hist(): データの分布を確認するヒストグラム。boxplot(): サンプル間の分布を比較し、外れ値を特定するボックスプロット。
例1: ベースRを使ったヒストグラムと散布図の作成
# サンプルデータ作成
set.seed(123)
data <- rnorm(100)
# ヒストグラム
hist(data, main = "Histogram of Data", xlab = "Value", col = "lightblue")
# 散布図
x <- rnorm(100)
y <- 2*x + rnorm(100)
plot(x, y, main = "Scatter Plot", xlab = "X", ylab = "Y")| グラフ名 | 対象となる構造 | 使用場面 | 関数名 | 入力データの構造・形式 | 主な引数(データ、スケール、ラベル、軸、色、タイトル) |
|---|---|---|---|---|---|
| 散布図 | 2変数(連続変数) | 変数間の関係性を確認する | plot() |
ベクトル、データフレーム(wide) | x, y(データ), xlim, ylim(スケール),
xlab, ylab(軸), col(色),
main(タイトル) |
| ヒストグラム | 1変数(連続変数) | データの分布を確認する | hist() |
ベクトル | x(データ), breaks(スケール),
xlab, ylab(軸), col(色),
main(タイトル) |
| ボックスプロット | 1変数または2変数(カテゴリ) | データの範囲、中央値、外れ値を確認する | boxplot() |
ベクトル、データフレーム(long) | x, y(データ), ylim(スケール),
xlab, ylab(軸), col(色),
main(タイトル) |
| バープロット | カテゴリデータ | カテゴリごとのカウントを視覚化 | barplot() |
テーブル、行列 | height(データ), ylim(スケール),
xlab, ylab(軸), col(色),
main(タイトル) |
| ペアプロット | 多変数(連続変数) | 変数間の関係性を一度に確認 | pairs() |
データフレーム(wide) | x(データ), xlim, ylim(スケール),
labels(ラベル), pch(色),
main(タイトル) |
| 線グラフ | 2変数(連続変数、時間変数) | 時系列データのトレンドを確認 | plot() |
ベクトル | x, y(データ), xlim, ylim(スケール),
xlab, ylab(軸), col(色),
main(タイトル) |
| パイチャート | カテゴリデータ | カテゴリごとの割合を視覚化 | pie() |
ベクトル、テーブル | x(データ), labels(ラベル),
col(色), main(タイトル) |
| ヒートマップ | 行列(多変数) | 行列形式のデータのパターンを視覚化 | heatmap() |
行列 | x(データ), scale(スケール),
xlab, ylab(軸), col(色),
main(タイトル) |
| 棒グラフ | カテゴリデータ | カテゴリごとの数値比較 | barplot() |
数値ベクトル、行列 | height(データ), ylim(スケール),
xlab, ylab(軸), col(色),
main(タイトル) |
高度な可視化パッケージによる詳細なカスタマイズ
ここではggplot2を中心に説明する。ggplot2は、高度にカスタマイズ可能な可視化ツールを提供し、発表や論文に使える品質のグラフを作成できる。また、ggplot2はレイヤーベースのアプローチを取っており、軸ラベルや色分け、凡例の位置などを細かく調整可能である。
1. ggplotにおける可視化の考え方
ggplot2 は、Grammar of
Graphics(可視化の文法)に基づいてデータを可視化するためのパッケージである。ここでの「文法」とは、可視化を構成する基本要素を組み合わせることで、多様なグラフを作成できるという考え方である。この文法に従ってデータを視覚化することで、データそのものの構造やパターンを強調した効果的な可視化を実現できる。
Grammar of Graphicsの基本要素
Grammar of
Graphics(可視化の文法)とは、データ可視化を一貫した方法で記述するための概念的なフレームワークである。この考え方は、Leland
Wilkinsonが提唱したもので、データをグラフィカルに表現する際に、複数の要素を組み合わせて「文法」として表現できるという考えに基づいている。ggplot2はこの文法に基づいて設計されたRのパッケージで、データを直感的かつ柔軟に可視化できる強力なツールである。Grammar
of
Graphicsは、データ可視化を以下のような主要要素に分解して説明する。これらの要素を組み合わせることで、任意の可視化を構築できる。
データ(Data)
グラフに表示する元となるデータセット。ggplot()の第一引数に指定する。審美的マッピング(Aesthetics Mapping, aes)
データのどの変数を、どの視覚的な要素(位置、色、大きさなど)に対応させるかを指定する。これにより、データの特定の側面を強調できる。例えば、x軸・y軸の位置や、色、形、サイズなどがある。aes(x = variable1, y = variable2, color = group)幾何オブジェクト(Geometries)
グラフの形状を定義する要素です。どの種類のグラフ(散布図、ヒストグラム、棒グラフなど)を描画するかを決定する。たとえば、geom_point()は散布図、geom_bar()は棒グラフを表す。geom_point() # 散布図 geom_bar() # 棒グラフスケール(Scales)
審美的マッピングで設定したデータを、どのようにスケール(変換)して視覚的に表現するかを指定する。軸の範囲、色の範囲、サイズの範囲などを調整する。scale_x_continuous()やscale_color_manual()などが使われる。scale_x_continuous(limits = c(0, 10)) # x軸のスケールを設定統計変換(Statistics)
データを集計、変換、あるいは計算して表示する方法を指定する。stat_summary()やstat_smooth()などが使われ、データのトレンドや要約統計量を表示する際に使われる。geom_smooth(method = "lm") # 線形回帰線を追加座標系(Coordinate Systems)
データの描画に使用する座標系を指定する。通常はcoord_cartesian()で直交座標系を使用するが、極座標なども選択可能である。coord_polar() # 極座標で描画ファセット(Facets)
データをグループ化し、複数のプロットに分割して表示する機能である。facet_wrap()やfacet_grid()を使うことで、グループごとに異なるサブプロットを自動的に作成できる。facet_wrap(~ group) # グループごとに分割して描画
ggplot2での可視化の流れ
まず、基本構文を先に示す。構造のみに着目すると、複数の関数が+で繋げられている。ggplot2では、この繋ぐ操作によって、可視化の文法(つまり各要素の組み合わせ)を実装している。
ggplot(data, aes(x = ..., y = ...)) +
geom_<plot_type>(aes(...)) +
labs(title = "Plot Title", x = "X-axis label", y = "Y-axis label") +
theme(...)さらに可視化の文法に従って、グラフを作成していくプロセスをみていこう。
データ形式の変換と可視化
データをlong形式またはwide形式に整形する。
dataset <- pivot_longer(wide_dataset)dataset <- pivot_wider(long_dataset)データを指定
描画するためのデータセットを指定する。ggplot(data = dataset)美学マッピングを指定
どの変数をx軸、y軸、色などに対応させるかを指定する。ggplot(data = dataset, aes(x = var1, y = var2, color = group))幾何オブジェクトを指定
散布図や棒グラフなど、どの形式でデータを描画するかを指定する。+ geom_point()スケールやテーマを設定
軸の範囲やカラースケール、テーマなど、グラフの外観を調整する。+ scale_x_continuous(limits = c(0, 10)) + theme_minimal()タイトルやラベルを設定
グラフにタイトルや軸ラベルを追加する。+ ggtitle("Title") + xlab("X-axis Label") + ylab("Y-axis Label")
例1: 散布図を作成する流れ
# ggplotによる散布図の作成例
library(ggplot2)
# サンプルデータ作成
data <- data.frame(x = rnorm(100), y = rnorm(100), group = rep(1:2, each = 50))
# 散布図を作成
ggplot(data, aes(x = x, y = y, color = as.factor(group))) + # データと美学マッピング
geom_point() + # 幾何オブジェクト
ggtitle("Scatter Plot Example") + # タイトル
xlab("X-axis") + ylab("Y-axis") + # ラベル
theme_minimal() # スケール・テーマこの例では、データの変数xとyを散布図の軸に対応させ、groupによって色分けしている。geom_point()を使用して散布図の点をプロットし、タイトルや軸ラベルも追加している。
long形式の重要性
ggplot2では、データの列ごとに異なる美学マッピング(aesthetic
mapping)を行いたいとき、データがlong形式であることが重要になる。例えば、同じサンプルに対して異なる条件(時間、治療、実験条件など)ごとに数値を比較する際や、複数の変数に対して色や形などを割り当てる場合、long形式のデータが適している。
生物学的データ解析において、定量データの最初の形式は、通常、wide形式で提供されることが多い。これは、行に特徴量(例えば、遺伝子やタンパク質)が並び、列にはサンプルや細胞が並んでいる形式であり、定量的な測定値が各セルに対応する。この形式は、計算や統計解析に便利である一方で、ggplot2による可視化には適さない場合がある。
一方で、発現変動解析などの解析パッケージによる出力結果では、しばしばp値や対数比変動、統計的有意性といった情報が、条件や群ごとに整理された形式で出力されることが多い。これらの出力結果は、long形式に近い構造を持っているか、あるいは少なくともlong形式に変換しやすい。
さらに、アッセイ情報に紐づくメタ情報(例えば、サンプルが属する条件や実験群)やアノテーション情報(例えば、遺伝子シンボル、GOカテゴリなど)も、可視化において重要な役割を果たす。これらの情報もまた、データの審美的属性(色、形、線の種類など)に対応する要素として使われる。メタ情報やアノテーション情報は、必ずしもwide形式では提供されず、しばしばlong形式、あるいはそれらが混在した形で提供される。
このような状況では、解析オブジェクト(SummarizedExperiment
や SingleCellExperiment
など)や解析結果から必要なデータを取り出し、適切にフィルタリングしてlong形式へ変換することが求められる。これにより、ggplot2による効果的な可視化が可能になる。例えば、定量データ(発現値)をlong形式に変換し、条件やアノテーション情報を結合したうえで、色や形にマッピングして視覚的に意味のあるグラフを生成することができる。
ggplot2による詳細な可視化一覧表
| グラフ名 | 対象となる構造 | 使用場面 | 関数名 | 入力データの構造・形式 | 主な引数(データ、スケール、ラベル、軸、色、タイトル) |
|---|---|---|---|---|---|
| 散布図 | 2変数(連続変数) | 変数間の関係性を視覚的に表現 | geom_point() |
データフレーム(long) | aes(x, y)(データ),
scale_x_continuous(), scale_y_continuous()(スケール),
labs(x, y)(軸), aes(color)(色),
ggtitle()(タイトル) |
| ヒストグラム | 1変数(連続変数) | データの分布を視覚的に表現 | geom_histogram() |
データフレーム(long) | aes(x)(データ),
scale_x_continuous()(スケール),
labs(x, y)(軸), aes(fill)(色),
ggtitle()(タイトル) |
| ボックスプロット | 1変数または2変数(カテゴリ) | カテゴリ間の分布の比較を行う | geom_boxplot() |
データフレーム(long) | aes(x, y)(データ),
scale_y_continuous()(スケール),
labs(x, y)(軸), aes(fill)(色),
ggtitle()(タイトル) |
| バープロット | カテゴリデータ | カテゴリごとの比較 | geom_bar() |
データフレーム(long) | aes(x)(データ),
scale_y_continuous()(スケール),
labs(x, y)(軸), aes(fill)(色),
ggtitle()(タイトル) |
| ペアプロット | 多変数(連続変数) | 変数間の関係性をペアで確認 | GGally::ggpairs() |
データフレーム(wide) | aes()(データ), axis.ticks(スケール),
labs()(軸), aes(color)(色),
ggtitle()(タイトル) |
| 線グラフ | 2変数(連続変数、時間変数) | 時系列データや連続変数のトレンドを可視化 | geom_line() |
データフレーム(long) | aes(x, y)(データ),
scale_x_continuous(), scale_y_continuous()(スケール),
labs(x, y)(軸), aes(color)(色),
ggtitle()(タイトル) |
| バイオリンプロット | 1変数または2変数(カテゴリ) | 分布の形状を詳細に確認 | geom_violin() |
データフレーム(long) | aes(x, y)(データ),
scale_y_continuous()(スケール),
labs(x, y)(軸), aes(fill)(色),
ggtitle()(タイトル) |
| ヒートマップ | 行列(多変数) | 行列形式のデータを可視化 | geom_tile() |
データフレーム(long) | aes(x, y, fill)(データ),
scale_fill_gradient()(スケール),
labs(x, y)(軸), ggtitle()(タイトル) |
| ファセットプロット | 多変数 | グループごとのプロット分割 | facet_wrap() |
データフレーム(long) | aes(x, y)(データ),
facet_wrap(~variable)(スケール),
labs(x, y)(軸), ggtitle()(タイトル) |
| PCA・UMAPプロット | 高次元データ | 次元削減後のデータを可視化 | geom_point() |
データフレーム(wide/long) | aes(x, y)(データ),
scale_x_continuous(), scale_y_continuous()(スケール),
labs(x, y)(軸), aes(color)(色),
ggtitle()(タイトル) |
演習:ggplot2を使った実際の解析場面を想定した可視化
以下の演習では、SummarizedExperimentオブジェクトを基に、特定の遺伝子の発現量を可視化したり、発現変動解析の結果を可視化することを通じて、ggplot2を使ったさまざまな可視化手法を学ぶ。
演習1: 特定遺伝子の発現量を条件間で比較
- 目的: サンプルごとの特定遺伝子の発現量を、条件(例えば治療群とコントロール群)で比較するプロットを作成する。
- 手順:
特定遺伝子のデータを抽出し、
ggplot2を使って箱ひげ図(ボックスプロット)を作成する。
# パッケージをロード
library(SummarizedExperiment)
library(ggplot2)
# サンプルデータ作成
counts <- matrix(rpois(1000, lambda = 10), ncol = 10)
colData <- DataFrame(condition = factor(rep(c("Control", "Treatment"), each = 5)))
se <- SummarizedExperiment(assays = list(counts = counts), colData = colData)
# 特定の遺伝子(例: gene1)の発現量を抽出
gene1_expr <- assay(se)["gene1", ]
gene1_data <- data.frame(Sample = colnames(se), Expression = gene1_expr, Condition = colData(se)$condition)
# 箱ひげ図を作成
ggplot(gene1_data, aes(x = Condition, y = Expression, fill = Condition)) +
geom_boxplot() +
ggtitle("Gene1 Expression by Condition") +
xlab("Condition") +
ylab("Expression Level") +
theme_minimal()演習2: 薬剤濃度依存の発現変化プロット
- 目的: 各サンプルにおける薬剤濃度に依存した発現変化を可視化する。
- 手順: 濃度ごとにサンプルをグループ化し、発現量の変化を折れ線グラフでプロットする。
# サンプルデータ作成
set.seed(123)
counts <- matrix(rpois(1000, lambda = 10), ncol = 10)
concentration <- c(0, 0.1, 1, 10, 50, 0, 0.1, 1, 10, 50)
colData <- DataFrame(concentration = concentration)
se <- SummarizedExperiment(assays = list(counts = counts), colData = colData)
# 特定遺伝子の発現量と濃度データを抽出
gene_expr <- assay(se)["gene2", ]
gene_data <- data.frame(Sample = colnames(se), Expression = gene_expr, Concentration = colData(se)$concentration)
# 薬剤濃度依存の発現変化プロット
ggplot(gene_data, aes(x = Concentration, y = Expression)) +
geom_point() +
geom_line() +
ggtitle("Gene2 Expression Change by Drug Concentration") +
xlab("Concentration (µM)") +
ylab("Expression Level") +
theme_minimal()演習3: 異常サンプルを散布図でラベル付き表示
- 目的: 発現データの中で異常なサンプルを散布図で可視化し、異常サンプルにラベルを付けて表示する。
- 手順: 散布図を作成し、閾値を超えた異常なサンプルにラベルを付けて表示する。
# サンプルデータ作成
set.seed(123)
counts <- matrix(rnorm(1000, mean = 10, sd = 2), ncol = 10)
colData <- DataFrame(condition = factor(rep(c("Control", "Treatment"), each = 5)))
se <- SummarizedExperiment(assays = list(counts = counts), colData = colData)
# 発現データの散布図
gene_expr <- assay(se)["gene3", ]
gene_data <- data.frame(Sample = colnames(se), Expression = gene_expr, Condition = colData(se)$condition)
# 異常サンプル(例えば、発現レベルが20を超えるサンプル)にラベルを付けて表示
ggplot(gene_data, aes(x = Condition, y = Expression)) +
geom_point() +
geom_text(aes(label = ifelse(Expression > 20, Sample, "")), vjust = -1) +
ggtitle("Scatter Plot with Outliers Labeled") +
xlab("Condition") +
ylab("Expression Level") +
theme_minimal()演習4: 発現変動解析の結果からボルケーノプロットを作成
- 目的: 発現変動解析の結果テーブルからボルケーノプロットを作成する。
- 手順: DESeq2の解析結果をもとに、ボルケーノプロットを作成し、統計的に有意な遺伝子を強調する。
# パッケージをロード
library(DESeq2)
library(ggplot2)
# サンプルデータ作成
set.seed(123)
dds <- makeExampleDESeqDataSet()
# 発現変動解析を実行
dds <- DESeq(dds)
res <- results(dds)
# NAの補完
res$log2FoldChange[is.na(res$log2FoldChange)] <- 0
res$pvalue[is.na(res$pvalue)] <- 1
# ボルケーノプロットの作成
ggplot(res, aes(x = log2FoldChange, y = -log10(pvalue))) +
geom_point(alpha = 0.5) +
geom_hline(yintercept = -log10(0.05), linetype = "dashed", color = "red") +
ggtitle("Volcano Plot of Differential Gene Expression") +
xlab("Log2 Fold Change") +
ylab("-Log10 P-value") +
theme_minimal()演習5: wide形式からlong形式への変換と可視化
目的:
データがwide形式で保存されている場合、それをlong形式に変換してから可視化する。特定の遺伝子発現量をサンプルごとに比較し、条件間での発現量の違いを可視化する。
手順:
pivot_longer()関数を使って、wide形式のデータをlong形式に変換する。ggplot2でボックスプロットを作成して、条件ごとの遺伝子発現量を比較する。
# パッケージをロード
library(tidyr)
library(ggplot2)
# サンプルデータ作成 (wide形式)
counts <- data.frame(
Gene = paste0("Gene", 1:5),
Sample1 = rpois(5, 10),
Sample2 = rpois(5, 12),
Sample3 = rpois(5, 14),
Sample4 = rpois(5, 16),
Sample5 = rpois(5, 18),
Condition = factor(c("Control", "Control", "Treatment", "Treatment", "Control"))
)
# データを確認 (wide形式)
print(counts)
# wide形式からlong形式へ変換
long_counts <- pivot_longer(counts, cols = starts_with("Sample"),
names_to = "Sample", values_to = "Expression")
# long形式データの確認
print(long_counts)
# 条件ごとのボックスプロットを作成
ggplot(long_counts, aes(x = Condition, y = Expression, fill = Condition)) +
geom_boxplot() +
ggtitle("Gene Expression by Condition") +
xlab("Condition") +
ylab("Expression Level") +
theme_minimal()演習6: 複数遺伝子の発現変動解析 (wideからlongへの変換)
目的:
複数の遺伝子について、条件間での発現量を比較するために、wide形式のデータをlong形式に変換し、条件ごとの発現量を可視化する。
手順:
- wide形式の遺伝子発現データを用意。
pivot_longer()でデータをlong形式に変換。ggplot2で条件ごとの発現量をボックスプロットで表示。
# パッケージをロード
library(tidyr)
library(ggplot2)
# サンプルデータ作成 (wide形式: 複数遺伝子の発現量)
rna_counts <- data.frame(
Gene = paste0("Gene", 1:5),
Control_Sample1 = rnorm(5, mean = 100, sd = 15),
Control_Sample2 = rnorm(5, mean = 102, sd = 14),
Treatment_Sample1 = rnorm(5, mean = 120, sd = 18),
Treatment_Sample2 = rnorm(5, mean = 122, sd = 17)
)
# wide形式からlong形式へ変換
long_rna_counts <- pivot_longer(rna_counts, cols = -Gene,
names_to = c("Condition", "Sample"),
names_sep = "_", values_to = "Expression")
# 発現量の可視化
ggplot(long_rna_counts, aes(x = Condition, y = Expression, fill = Condition)) +
geom_boxplot() +
facet_wrap(~ Gene, scales = "free_y") +
ggtitle("Gene Expression Comparison by Condition") +
xlab("Condition") +
ylab("Expression Level") +
theme_minimal()演習7: 時系列データのlong形式変換と可視化
目的:
時系列データをlong形式に変換し、各遺伝子の発現量が時間とともにどのように変化するかを可視化する。
手順:
- wide形式で保持されている時間ごとの発現データを
pivot_longer()でlong形式に変換。 ggplot2で各遺伝子の時系列プロットを作成。
# パッケージをロード
library(tidyr)
library(ggplot2)
# サンプルデータ作成 (wide形式: 時系列データ)
time_series <- data.frame(
Gene = paste0("Gene", 1:5),
Time0 = rnorm(5, mean = 100, sd = 10),
Time1 = rnorm(5, mean = 105, sd = 12),
Time2 = rnorm(5, mean = 110, sd = 14),
Time3 = rnorm(5, mean = 115, sd = 13)
)
# wide形式からlong形式へ変換
long_time_series <- pivot_longer(time_series, cols = -Gene,
names_to = "Time", values_to = "Expression")
# 時系列プロットを作成
ggplot(long_time_series, aes(x = Time, y = Expression, group = Gene, color = Gene)) +
geom_line() +
geom_point() +
ggtitle("Gene Expression Over Time") +
xlab("Time") +
ylab("Expression Level") +
theme_minimal()演習8: 遺伝子間の相関関係の可視化 (wideからlongへの変換)
目的:
複数の遺伝子間の相関を計算し、その相関行列をlong形式に変換してヒートマップを作成する。
手順:
- 遺伝子発現データから相関行列を計算。
melt()を用いて相関行列をlong形式に変換。ggplot2でヒートマップを作成。
# パッケージをロード
library(tidyr)
library(ggplot2)
library(reshape2)
# サンプルデータ作成 (wide形式: 複数遺伝子の発現量)
gene_data <- data.frame(
Gene1 = rnorm(10, mean = 100, sd = 15),
Gene2 = rnorm(10, mean = 110, sd = 20),
Gene3 = rnorm(10, mean = 120, sd = 25),
Gene4 = rnorm(10, mean = 130, sd = 30)
)
# 遺伝子間の相関行列を計算
cor_matrix <- cor(gene_data)
# 相関行列をlong形式に変換
long_cor_matrix <- melt(cor_matrix)
# ヒートマップで相関を可視化
ggplot(long_cor_matrix, aes(x = Var1, y = Var2, fill = value)) +
geom_tile() +
scale_fill_gradient2(low = "blue", high = "red", mid = "white", midpoint = 0) +
ggtitle("Gene Correlation Heatmap") +
xlab("Gene") +
ylab("Gene") +
theme_minimal()